Last updated: March 05, 2012

Researchers use genomics to differentiate two ovarian cancer subtypes
Researchers use genomics to differentiate two ovarian cancer subtypes
By Raymond MacDougall
Associate Communications Director for Intramural Research
 |
"This study takes a new look at ovarian cancer using genomic tools and analysis methods," said Laura Elnitski, Ph.D., senior author and NHGRI investigator. "We recognize clear patterns of difference from one subtype of this cancer to another, which is the key to making more accurate clinical diagnoses and, ultimately, offering individualized cancer treatments."
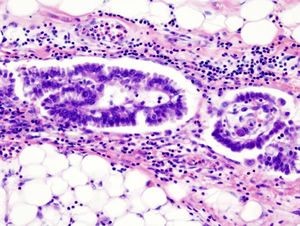
Ovarian cancer, one of women's most serious health challenges, is characterized by uncontrolled and abnormal cell growth that results from the accumulation of mutations in the normal DNA sequence. These mutations can create defects in genes and proteins that healthy genes would otherwise produce or disturb a specific biochemical process — DNA methylation — that toggles genes on or off. Each year, more than 21,000 women are diagnosed with ovarian cancer in the United States. Because it has few early warning signs, the five-year survival rate is less than 30 percent, and each year 125,000 women around the globe die from this disease.
With colleagues from the George Washington University School of Public Health and Health Services in Washington, D.C., Rush University in Chicago, and the University of Pittsburgh Medical School, NHGRI researchers embarked on one of the broadest comparative studies of DNA methylation in gynecologic cancers. They examined more than 27,500 methylation sites in the promoter region of almost 14,500 genes. A promoter region is a segment of DNA that is important in controlling the expression of a gene.
Ovarian cancer has a complex makeup with at least four distinct subtypes, each characterized by a unique appearance under a microscope. The researchers analyzed DNA methylation patterns for two subtypes. They focused on serous, the most common form of the disease, and endometrioid, another aggressive type that parallels characteristics of tumors found in the lining of the uterus. Though treatment options have broadened and become increasingly sophisticated for cancer of the ovary, serous and endometrial subtypes are regarded and treated as a single disease.
The researchers compared serous and endometrioid ovarian tumors with healthy tissues from the fallopian tube and uterine lining, as well as with tumors of the uterus. Further, they drew upon methylation data from serous tumors and healthy fallopian tube tissue from The Cancer Genome Atlas, a consortium sponsored jointly by the NHGRI and the National Cancer Institute at the National Institutes of Health.
The researchers looked for methylation patterns to take shape in their genomic data using computational analyses of thousands of points of data that resulted from profiling a large number of tissue types and targeted genes. They found patterns of altered methylation coincided with the different cancer types.
For instance, endometrioid tumors showed extensive accumulation and loss of methylation in the gene promoter regions that reflects defective maintenance of the pattern. Similar accumulations of DNA methylation have been seen in colon and liver cancers and are known as a methylator phenotype.
Endometrioid tumors carried very similar alterations, regardless of whether they arose in the ovary or uterus. In contrast, serous tumors had methylation patterns more like normal cells and levels of methylation that were less consistent than endometrioid tumors.
The study provides a foundation for deeper understanding of uterine and serous tumors at the molecular level, according to the authors. This understanding will be useful for future design and development of individualized diagnostic and treatment applications, which utilize genomic and genetic information of each patient.
Posted: March 5, 2012