Last updated: February 22, 2012

Developing a Haplotype Map of the Human Genome for Finding Genes Related to Health and Disease
Developing a Haplotype Map of the Human Genome
for Finding Genes Related to Health and Disease
Washington, D.C.
July 18-19, 2001
Introduction
So far about 2.4 million DNA sequence variants (single nucleotide polymorphisms or SNPs) have been discovered in the human genome, and millions more exist. These variants will be most useful for discovering genes related to health and disease if their organization along chromosomes, the haplotype structure, is known. Technology is just reaching the point that haplotype maps of blocks of SNPs along chromosomes can be developed.
On July 18-19, 2001, the National Institutes of Health (NIH) held a meeting in Washington, D.C., to discuss how haplotype maps could be used for finding genes contributing to disease; the methods for constructing such maps; the data about haplotype structure in populations; the types of populations and samples that might be considered for a map; the ethical issues, including those related to studying genetic variation in identified populations; and how such a project could be organized. The goal was to resolve some issues and to set up procedures for resolving others.
There were 165 attendees, including human geneticists, population geneticists, anthropologists, pharmaceutical and biotech industry scientists, social scientists, ethicists, representatives from various communities and disease groups, administrators from many NIH institutes and international funding agencies and journalists.
Background: Genetic Variation and Its Use for Mapping Genes Contributing to Disease
Recently technology has become available to study the extent and pattern of human genetic variation on a large scale, and to use this variation to find the genes that contribute to disease. The information summarized here was not presented at the meeting but provides the background for understanding the importance and use of a haplotype map.
Rationale for finding genes contributing to disease
The goal of much genetic research is to find genes that contribute to disease. Finding these genes should allow an understanding of the disease process, so that methods for preventing and treating the disease can be developed. For diseases with a relatively straightforward genetic basis, the single-gene disorders, current methods are usually sufficient to find the genes involved. Most people, however, do not have single-gene disorders, but develop common diseases such as heart disease, stroke, diabetes, cancers or psychiatric disorders, which are affected by many genes and environmental factors. The genetic contribution to these diseases is not clear, but many researchers consider common variants to be important, the Common-Disease/Common-Variant theory.
Definition of a Single Nucleotide Polymorphism, SNP
A SNP is a site in the DNA where different chromosomes differ in the base they have. For example, 30 percent of the chromosomes may have an A, and 70 percent may have a G. These two forms, A and G, are called variants or alleles of that SNP. An individual may have a genotype for that SNP that is AA, AG, or GG.
Number of SNPs
When chromosomes from two random people are compared, they differ at about one in 1000 DNA sites. Thus when two random haploid genomes are compared, or all the paired chromosomes of one person are compared, there are about three million differences. When more people are considered, they will differ at additional sites. The number of DNA sites that are variable (SNPs) in humans is unknown, but there are probably between 10 and 30 million SNPs, about one every 100 to 300 bases. Of these SNPs, perhaps four million are common SNPs, with both alleles of each SNP having a frequency above 20 percent.
How SNPs are used to find genes contributing to disease
Some SNP alleles are the actual functional variants that contribute to the risk of getting a disease. Individuals with such a SNP allele have a higher risk for that disease than do individuals without that SNP allele. Most SNPs are not these functional variants, but are useful as markers for finding them. To find the regions with genes that contribute to a disease, the frequencies of many SNP alleles are compared in individuals with and without the disease. When a particular region has SNP alleles that are more frequent in individuals with the disease than in individuals without the disease, those SNPs and their alleles are associated with the disease. These associations between a SNP and a disease indicate that there may be genes in that region that contribute to the disease.
The use of haplotypes
A haplotype is the set of SNP alleles along a region of a chromosome. Theoretically there could be many haplotypes in a chromosome region, but recent studies are typically finding only a few common haplotypes. Consider the example below, of a region where six SNPs have been studied; the DNA bases that are the same in all individuals are not shown. The three common haplotypes are shown, along with their frequencies in the population. The first SNP has alleles A and G; the second SNP has alleles C and T. The four possible haplotypes for these two SNPs are AC, AT, GC, and GT. However, only AC and GT are common; these SNPs are said to be highly associated with each other.
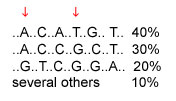 The cost of genotyping is currently too high for whole-genome association studies that would look at millions of SNPs across the entire genome to see which SNPs are associated with disease. If a region has only a few haplotypes, then only a few SNPs need to be typed to determine which haplotype a chromosome has and whether the region is associated with a disease. In the example below, typing two SNPs is all that is needed to distinguish among the three common haplotypes. The two SNPs indicated by arrows are one pair of several possible pairs of SNPs that distinguish among the three common haplotypes.
The cost of genotyping is currently too high for whole-genome association studies that would look at millions of SNPs across the entire genome to see which SNPs are associated with disease. If a region has only a few haplotypes, then only a few SNPs need to be typed to determine which haplotype a chromosome has and whether the region is associated with a disease. In the example below, typing two SNPs is all that is needed to distinguish among the three common haplotypes. The two SNPs indicated by arrows are one pair of several possible pairs of SNPs that distinguish among the three common haplotypes.
Most SNP variation is within all groups
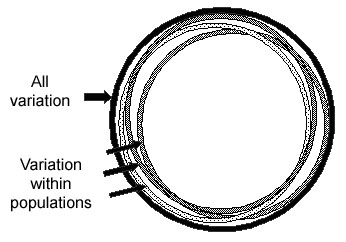 For most SNPs, any population has individuals of all possible genotypes for a SNP, but populations differ in the frequencies of individuals with each of the different genotypes. About 85 percent of human SNP variation is within all populations, and about 15 percent is between populations, as shown in the figure below. Thus two random individuals within a village are almost as different in their SNP alleles as any two random individuals from anywhere in the world. Although a small proportion of SNPs have alleles that are common in some groups but rare in others, most SNP alleles that are common in one group will be common in other groups. Under the Common-Disease/Common-Variant theory, common variants that contribute to a disease in one group will also contribute to the disease in other groups, although the amount of the contribution may vary.
For most SNPs, any population has individuals of all possible genotypes for a SNP, but populations differ in the frequencies of individuals with each of the different genotypes. About 85 percent of human SNP variation is within all populations, and about 15 percent is between populations, as shown in the figure below. Thus two random individuals within a village are almost as different in their SNP alleles as any two random individuals from anywhere in the world. Although a small proportion of SNPs have alleles that are common in some groups but rare in others, most SNP alleles that are common in one group will be common in other groups. Under the Common-Disease/Common-Variant theory, common variants that contribute to a disease in one group will also contribute to the disease in other groups, although the amount of the contribution may vary.
The Meeting
At the meeting there was discussion of recent data related to haplotype maps, and what a haplotype map might look like. Since haplotypes and associations of SNPs with disease are population phenomena, some of the discussion focused on complexities in sampling human populations. Much of the discussion concerned the information that could be gained by identifying the populations contributing samples for a haplotype map, and the risks and benefits to populations of such identification. The meeting ended with discussion of various aspects of a haplotype map project, including what issues would need more discussion in working groups after the meeting. The main points of the discussions are summarized here.
The Pattern of Genetic Variation and Association Among Genes
Factors that affect the frequencies of alleles and haplotypes in populations:
- Biological factors: Haplotype and allele frequencies are affected by cellular-level processes such as mutation, recombination, and gene conversion, as well as by population-level processes such as natural selection against alleles that contribute to disease. When genes are close together and associated, then selection that changes the frequency of an allele at one gene results in similar changes in the frequencies of alleles at other genes on the same haplotype.
Recombination is the major process that breaks down the associations between SNPs. It is unclear whether haplotype block boundaries are due to recombination hotspots, or are simply the result of recombination events that happened to occur there. If the blocks are due to hotspots, then perhaps they will be common across populations. If the blocks are due to regular recombination events, then populations may or may not share them, depending on how long ago the recombination events occurred. When large chromosomal regions are examined, the regions with high association have less recombination and less genetic variation.
- Demographic and social factors: Haplotype and allele frequencies are also affected by population history factors such as population size, bottlenecks or expansions of population size, founder effects, isolation of a population or admixture between populations, and patterns of mate choice.
- Large variances: The many influences on haplotype frequencies result in large statistical variances for associations among different SNPs; all such studies find that associations vary a lot around the mean. Neighboring SNPs may not be associated, while distant SNPs may be associated, despite the average association declining with longer distances between SNPs. This variance means that more SNPs are needed to study associations than simply counting the blocks might indicate.
Extent of association among SNPs differs by chromosome region and by allele frequency
Some studies show that a measure of association, D', falls to half its possible maximum value at a distance between SNPs of about 50 to 80 kb, averaged over gene regions in European-derived populations. Some regions have strong associations over as much as one megabase. Among different chromosome regions there is about a fourfold range in the extent of associations. Rare SNP alleles are generally of more recent origin than common SNP alleles; recombination has had less time to break down associations around them so that rare alleles generally have associations over longer distances than do common alleles.
Extent of association among SNPs differs by population
Many studies show that the chromosomal distances that SNP associations extend are generally shorter for African populations, intermediate for European and Asian populations, and longer for American Indian populations, although there is variation among populations in the same geographic region. When groups of people from populations that differ in some allele frequencies marry and reproduce with each other, as has often happened with African-Americans and with Hispanics in the United States, associations are generated over longer chromosomal distances in the admixed group than in either parental group. Recently formed populations such as the Mennonites and Acadians also may have associations over longer chromosomal distances.
Common haplotypes are in all populations
The pattern of variation within and among populations for haplotype structure is just starting to be studied on a large scale. Recent studies show that the common haplotypes are found in all populations studied, and that the population-specific haplotypes are generally rare. African populations generally have more haplotypes than other populations, which generally have subsets of the African ones, due to the origins of other populations from ones that spread out of Africa.
Haplotype Block Structure as the Rationale for a Haplotype Map
Block pattern of haplotypes
Some recent studies found that haplotypes occur in a block pattern: the chromosome region of a block has just a few common haplotypes, followed by another block region also with just a few common haplotypes, with the longer-distance haplotypes showing a mixing of the haplotypes in the two blocks. Another description of this pattern is that the SNPs in a block are strongly associated with each other, but much less associated with other SNPs. Blocks range in size from about three kb to more than 150 kb. The majority of SNPs are organized in these blocks. Some recent data show that the blocks in a Yoruban population from Nigeria are generally the same ones as, but shorter than, those in two European-derived populations, although the data are limited and these conclusions are preliminary.
Using haplotype blocks to find chromosome regions associated with disease
Where blocks exist, they can be tested for association with a disease, using just a few SNPs per block. If the blocks are large, then a few SNPs in a region will indicate whether that region has genes related to a disease. If the blocks are small, then many SNPs will be needed to cover a region. Typing more SNPs than needed is a waste of resources; typing too few SNPs means that a disease association could easily be missed.
A haplotype map
A haplotype map would show the haplotype blocks and the SNPs that define them. A haplotype map thus would serve as a resource to increase the efficiency and comprehensiveness of the many other studies that will be done to relate genes to diseases.
Haplotype maps of different populations
To the extent that populations differ in their haplotype structure, it may be useful to study different populations during different stages of the process of finding disease genes. Studying populations with large haplotype blocks will be useful for initial association studies over the entire genome to find chromosome regions affecting a disease. Once these chromosome regions have been found, they can be studied in populations with small haplotype blocks in those regions, so the particular genes can be found more easily by being localized in small regions.
Sampling Human Haplotype Variation
Possible schemes to sample human haplotype variation
- Population sampling: Samples are chosen from particular identified populations, defined by ethnicity and geography.
- Grid sampling: Samples are chosen from particular geographic regions on a world grid.
- Proportional sampling: Samples are chosen from identified populations so that the entire sample has a known distribution, but the population identities of the individual samples are not kept. This scheme was used for the DNA Polymorphism Discovery Resource.
Studying one or multiple populations
Studying just one population would reveal the common haplotypes that are in all populations, and so the resulting haplotype map would be useful for all populations. Including only one, non-disadvantaged, population would also avoid some of the ethical issues raised by identifying populations. However, this approach would raise serious issues of justice, since only that population could receive the population-specific advantages of the haplotype map. There are also scientific reasons to include more than one population in a haplotype map: to add haplotypes that are not as common or that are more variable in frequency among populations; and to reveal regions that are similar or different in haplotype structure among populations. After the first few populations are included in a haplotype map, additional populations could still be added. The haplotype map should be developed so that it would be useful for mapping genes in any population.
Designating which population an individual belongs to, when choosing which individuals to sample
There are many ways that individuals could define which populations they belong to, such as their cultural affiliations or the geographic origins of their grandparents. Most populations have blurred boundaries, some more than others. Some people define themselves as members of several populations. Individuals or communities may emphasize some aspects of ancestry more than others, based on factors such as pride, shame, history of discrimination, or extent of knowledge. For a haplotype map, the purpose of designating an individual as belonging to a particular population is simply to make sure that most of that person's bi-parental lineages come from that particular population. Occasional differences between the population designations of individuals and their actual lineages would have little effect on a haplotype map, since the blocks are defined by the common haplotypes in a population contributing samples. The complexity in designating individuals as belonging to particular populations underscores the need for involving experts in the social sciences when developing a haplotype map.
Population consultation
Only a few populations would need to be included for the haplotype map to become a useful resource for individuals in all populations; there is no reason why any particular population should have to participate for the project to succeed. For any population that might be included in a haplotype map, there must be a process of community consultation to explain the purpose of the map and identify issues of concern to that population. This process would take time but would be necessary to educate both the population and the researchers. Particular populations may be sensitive to being exploited or to being left out. Issues may arise that require modifications to the consent process, the research protocol, the procedures of the sample repository, or the database. American Indian and Alaskan Native tribes are sovereign nations and have procedures for formally granting or withholding consent to research, which by law must be followed. Other populations are less well organized, making formal population consent unobtainable, but community consultation would still be needed, keeping in mind the multiple geographic scales and other complexities that characterize many populations. Procedures for consulting communities are outlined in an emerging literature and are under discussion at NIH.
Issues Associated with Identifying Samples by Population
Risks in identifying populations
Identifying the populations that contribute samples for a haplotype map could raise ethical risks. One risk is that any racial or ethnic identifiers used for the map would come to be reified as biological constructs, fostering a genetic essentialism in the way the map is interpreted and the categories understood. This essentialism could obscure the fluid nature of the "boundaries" between groups and the common genetic variation within all groups. Although the haplotype map would not have any individual medical information, another risk to the groups that participate could arise from later studies that use the haplotype map to find genes contributing to diseases; the participating groups could become more intensely studied, leading to the perception that their members are at high risk for diseases.
Benefits in identifying populations
Identifying individual samples as contributed by members of particular populations would be most useful scientifically. For each population, it would allow multiple sources of biomedical information to be combined. The contributing populations would gain the general benefits of the haplotype map as well as any additional benefits from studies of those particular populations. However, it is an open question how much less useful a haplotype map would be if population identifiers were omitted. Additional studies of population differences in haplotypes are needed to resolve this issue.
Describing the contributing populations
Regardless of whether the individual samples would be identified by their population of origin, any populations that contribute to a haplotype map must be described in a way that does not reinforce the mistaken perception that populations are genetically distinct, well-defined groups. Because people take in information most readily when it confirms their stereotypes, terms related to race and ethnicity must be used with precision, sensitivity, and care. Populations should be described as specifically as possible; for example, if a group of Chinese-Americans in Hawaii were studied, the population should not be labeled simply "Chinese." This specificity of description is crucial to minimize the risk of essentialist definitions of race, which assume that all individuals of a race are genetically similar.
Other Issues Raised by a Haplotype Map Project
Health priorities
Some communities lack even basic health care, so a haplotype map may be a low priority for them. Groups may feel that even if they participate in a haplotype project, not much attention would be paid to genetic diseases that primarily affect some of them but not members of other groups.
Sampling in the developed vs. the developing world
Including samples from developed countries and regions, such as the United States, Canada, Europe and Japan, might raise fewer human subjects concerns than would samples from developing countries without good IRB systems for overseeing research or strong biomedical research infrastructures.
Why not obtain the medical phenotypes of the sampled individuals when developing a haplotype map?
No phenotypic data, such as medical information, would be collected along with the samples. The haplotype map would be a resource for researchers trying to relate genetic variation to a wide range of disorders and traits. Only about 50 samples from each population would be needed to develop a haplotype map. Such a small sample would not be adequate to evaluate the many genetic and environmental factors that affect a disease. However, if the haplotype structure of the genome and the identifying SNPs were known, then researchers could use those SNPs in studies of individuals affected and not affected by a disease, matched to control for environmental factors, to track down the genes that contribute to the disease.
Elements of a Research Plan
The goal of a haplotype map should be medical
A haplotype map could be set up in many ways, to support various types of medical and biological research. It should be set up to best facilitate its use for relating genetic variation to disease.
How should a haplotype block be defined?
To compare studies, it will be important to develop a standard definition of a block, including the minimum frequencies of alleles for SNPs used, how similar haplotypes must be to be considered the "same" haplotype when figuring out which haplotypes are common, and how much of a drop in association defines the boundaries of blocks. Descriptions of the block structure of the genome would include distributions of the lengths of blocks, measures of the variability of blocks, the amount of coverage of the genome by blocks, and the proportion of haplotypes in common blocks; there are tradeoffs among these measures depending on the values of the defining parameters. Care will be needed when comparing studies using SNPs with different allele frequencies, with SNPs ascertained in different ways, and with different sample sizes for estimating associations.
Pilot projects, to help decide whether population identifiers are needed
Population differentiation for allele frequency is about 7 to 10 percent. However, currently little is known about how populations differ in their haplotypes. It will be important to find out whether the haplotype blocks are the same in different populations. Are differences in the extent of associations due to differences in block lengths, or to differences in the associations among neighboring blocks? How different are different populations from the same geographic region? How much information would be lost by removing population identifiers from samples? The first step would be to get more data, by sampling a small set of populations with different geographic origins. If these populations were similar, then it may be possible not to identify populations and still get a haplotype map that can be broadly useful. If the populations were different enough, then it might be necessary to identify the populations that contribute the samples. Projects already underway might be used to answer this question, or some pilot projects might be needed.
Number of populations
It was suggested that about 3 to 6 populations would be included in a haplotype map. The goal is to produce a tool that is broadly useful.
Samples from real populations
To obtain the most representative samples, it is important not to use samples of convenience, but to choose samples from real populations. The populations that contribute samples should be chosen based on the goals of the haplotype map, and the samples should be collected with appropriate population consultation and informed consent.
Common samples
Having a common set of samples that could be used by all research groups would allow comparisons among the results of different studies. Combining information across studies produces much more informative results than simply the sum of the results of separate studies.
SNP allele frequencies
Inclusion of SNPs spanning the range of SNP allele frequencies would be important. The length of associations among alleles may differ depending on the frequencies of the alleles. Also, a SNP allele provides the most power for an association study when its frequency matches that of a nearby allele contributing to a disease, and such alleles can be expected to span the range of frequencies.
A hierarchical approach for SNP density
It would be needlessly expensive to genotype all the individuals in a sample with a dense set of SNPs. A hierarchical approach makes more sense: start with a density of SNPs of perhaps one every 50 kb. For regions where such adjacent markers are strongly associated, these SNPs are sufficient and should be able to define blocks. For other regions, SNPs with a density of perhaps one every 10 kb could be examined, and so on until only regions with no block structure are left. Another type of hierarchical approach would be to start in the regions around genes.
What would define the endpoint of the project?
The goal of a haplotype map is to have sufficient SNPs so that researchers doing association studies could be sure that regions containing disease alleles have been found, and that regions not containing disease alleles can be excluded from further consideration. The map could be considered complete when more SNPs provide no more information about block structure, or when all common SNPs are included in the map or are highly associated with ones included.
Methods
Many technical issues still need to be worked out: the method for determining haplotypes; the types of samples needed, such as single chromosomes, individuals or families with a certain number of children; the number of samples for each population; and quality measures. The processes for consulting with populations and obtaining informed consent need to be developed.
Data analysis
Dealing with thousands to millions of SNPs, haplotypes, and haplotype blocks requires the development of better statistical methods of analysis to delineate blocks and to associate them with diseases. Better analytical methods are needed to model and understand the chromosomal and population processes that lead to the block structure observed.
Open data-sharing policy
Just as was done for the sequence produced by the Human Genome Project, providing rapid and complete data release to appropriate public databases would allow maximal benefit to be gained from haplotype data by allowing all researchers quick access to the data.
Coordination of data producers
A haplotype project would need coordination among the data producers, both large and small. The project should be international and open to all interested researchers.
Process for Planning
International project
An international steering committee should be formed. So far there is interest from the United States, Canada, the United Kingdom, France, Germany and Japan.
Two working groups
Some issues could be considered by one group; others could be considered by both.
- Population and ELSI Group: To identify the risks associated with a haplotype map project, including those associated with identifying populations; to consider how to minimize those risks; and to consider which types of populations should be considered for inclusion in a haplotype map.
- Methods Group: To consider the types of samples needed and how to create a haplotype map.
Name of the project
The public would have a hard time understanding and supporting a project named anything like The Haplotype Linkage Disequilibrium Association Map. A more understandable name is needed, as well as better ways to explain the project. It will also be important to communicate clearly what is understood about the complex relationships among genetics, culture, race and ethnicity.
Last Reviewed: February 22, 2012