Last updated: March 05, 2015

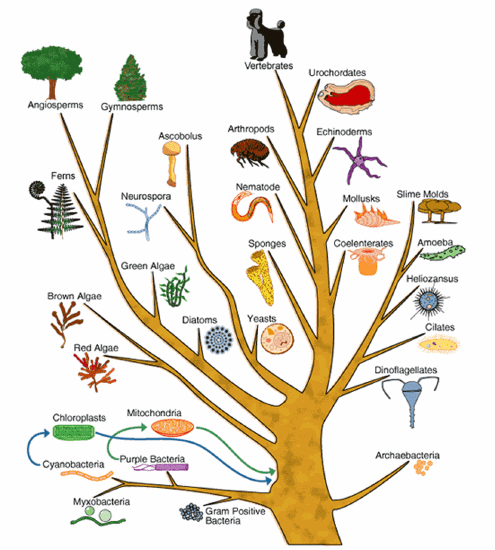
Bioinformatics: Finding Functions
Bioinformatics: Finding Functions
Once a nucleic acid or amino acid sequence has been assembled, bioinformatic analysis can be used to determine if the sequence is similar to that of a known gene. This is where sequences from model organisms are helpful. For example, let's say we have an unknown human DNA sequence that is associated with the disease cystic fibrosis. A bioinformatic analysis finds a similar sequence from mouse that is associated with a gene that codes for a membrane protein that regulates salt balance. It is a good bet that the human sequence also is part of a gene that codes for a membrane protein that regulates salt balance.
Determining the similarity of two sequences is not as easy as you might think. For example, it was recently reported that the genomes of humans and chimpanzees are 96 percent similar. What does this really mean?"
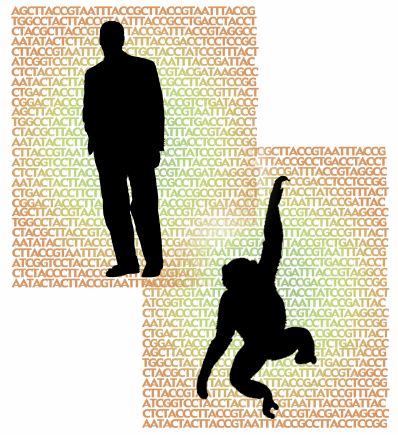
Consider the following two sequences:
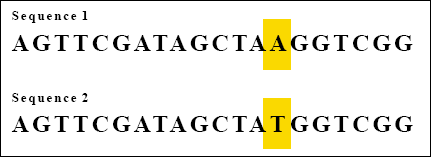
Each sequence consists of 20 bases. There is just one base difference between them. Because the two sequences match at 19 out 20 bases, we can say that the two sequences are 95 percent the same.
Now consider the following two DNA sequences:
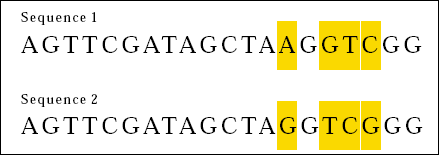
This time, 16 out of 20 bases match. We can say that the two sequences are 80 percent the same. Careful inspection however reveals another sort of similarity between Sequences 3 and 4.
If we align the sequences like this . . .

We see that the two sequences differ by just a missing base in Sequence 4 (or an added base to Sequence 3).
Does the deletion (or insertion) of a single base equal four base substitutions as suggested in this example? There is no simple answer to that question. When comparing sequences, we must be concerned not only with the quantity of the differences but the quality as well.
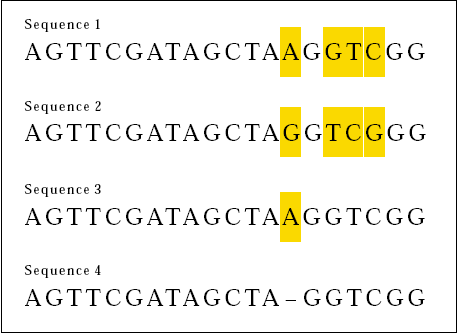
Scientists have written computer programs that can be used to see if a particular DNA sequence is similar to any others that are stored in a sequence database. One of the most popular such programs is called BLAST (Basic Local Alignment Search Tool). Using this program is somewhat like using a search engine on the Internet. The user provides the program with a biological sequence (when using BLAST) or a subject (when using a search engine). In each case, the program compares the input information to the information found in the database. The results are given with the most closely matching items (or sequences) listed first, followed by items (or sequences) that match less well.

Let's look at an example of a BLAST search. The input sequence that is being compared to others in the database is called the query sequence. In our example, the query is the short human DNA sequence listed below.

Once the query sequence is submitted, the BLAST program compares it, one-at-a-time, to every sequence in its database. Typically, the search results are displayed so that the query sequence is shown at the top and the matching sequences are listed below it. The listed sequence "hits" also may include links to relevant bibliographic information. The results from this search are shown below.

BLAST Search Terminology:
Sequence ID: A unique number used to identify the DNA sequence.
Description: Describes the species from which the sequence comes and the gene it is associated with (if any).
Query: Indicates how many bases are in the input (test) sequence.
Match: The amount of shading on each graphic indicates how well the query sequence matches the hit (or subject) sequence. Note, the shading does not compare the similarities between the whole genomes.
Expected (E) Value: Result of a mathematical calculation that describes the significance of a match. The lower the E value (closer to"0"), the better the match. An E value of less than 10-6 is a biologically significant match.