Last updated: March 05, 2015

Bioinformatics: Examining Variation
Bioinformatics: Examining Variation
The BLAST program compares a single input sequence, one at a time, to others in a sequence database. The results can provide clues as to the identity and function of the input sequence. Sometimes you may want to compare a number of different sequences, all at the same time to see where they are alike and where they are different. The CLUSTAL program was developed to produce such multiple alignments. CLUSTAL gets its name because it deals with clusters of sequences.
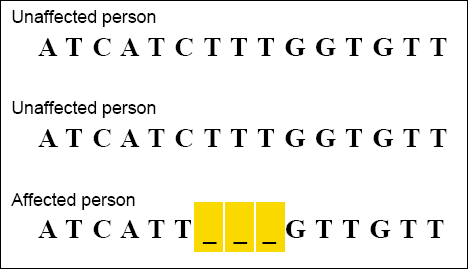
CLUSTAL alignments are sometimes used by scientists examining genetic variation within a population. For example, once a gene has been associated with a disease, scientists can use CLUSTAL to examine how the gene sequence varies among people with and without the disease. The example below shows a CLUSTAL alignment of DNA sequences from a portion of the gene associated with cystic fibrosis. The person affected by the disease is seen to be missing a three-base DNA sequence.
Multiple sequence alignments are also useful to scientists investigating the evolutionary relationships among species. For example, the CLUSTAL program can be used to align a series of related sequences from different species. Once the program has produced the best alignment for the sequences, another program can calculate the evolutionary relationships between them. These data can be used to construct a tree diagram showing the evolutionary relationships for that sequence among the various species.
