Last updated: July 31, 2012

New Findings Raise Questions About Process Used to Identify Experimental Drug for Rare Genetic Diseases

National Human Genome Research Institute
www.genome.gov
New Findings Raise Questions About Process Used to Identify Experimental Drug for Rare Genetic Diseases
 |
Bethesda, Md., Mon., Feb., 2, 2009 — A study by National Institutes of Health (NIH) researchers has revealed surprising new insights into the process used to initially identify an experimental drug now being tested in people with cystic fibrosis and muscular dystrophy. Researchers emphasized that the clinical implications of their findings are unclear, but said the results suggest more work may be needed to make sure the screening process to select promising agents was not flawed by its effects on a firefly enzyme used as a marker. The study was published today in the Proceedings of the National Academy of Sciences (PNAS).
Over the past several years, an experimental drug called PTC124 has generated excitement among those seeking treatments for inherited diseases caused by a type of genetic alteration that leads to production of abnormally short proteins. Scientists refer to such alterations as nonsense mutations. About 10 percent of cystic fibrosis cases and about 15 percent of Duchenne muscular dystrophy cases are thought to arise from nonsense mutations. In addition, nonsense mutations may affect a substantial portion of the approximately 25 million Americans suffering from other rare, genetic disorders.
The enzyme that makes fireflies glow, called luciferase, is widely used in biomedical experiments and in high-throughput screening often utilized to discover drugs. In a positive reaction, the tested material literally lights up.
PTC Therapeutics of South Plainfield, N.J., identified PTC124 through a cell-based screening system that used firefly luciferase to gauge the power of chemical compounds to enable cells with nonsense mutations to produce normal, full-length proteins. In the PTC screen, a particular group of test compounds elicited very bright signals from the firefly enzyme.
Company researchers interpreted the bright signals to mean that such compounds were highly active - that they had enabled the cellular machinery to efficiently read through nonsense mutations and increase the production of full-length, functional protein. One of those compounds was then optimized, also using firefly luciferase tests, to develop the experimental drug PTC124.
The news about PTC124 was particularly encouraging because previous research had suggested that restoring the ability to produce even low levels of full-length protein might ease symptoms of cystic fibrosis and possibly other diseases caused by nonsense mutations. The drug used in that previous work, however, an antibiotic called gentamicin, is difficult to administer and can lead to hearing loss and life-threatening kidney damage.
Additional testing in animals and healthy human volunteers showed that PTC124 seemed safe. Moreover, PTC124 was shown to increase the amount and function of critical proteins in animal and cell-culture models that do not rely on firefly luciferase as a marker, namely, the cystic fibrosis transmembrane conductance regulator protein in models of cystic fibrosis, and the dystrophin protein in models of Duchenne muscular dystrophy.
Based on those results, several years ago PTC Therapeutics launched clinical trials of an oral form of PTC124 for cystic fibrosis and Duchenne muscular dystrophy, diseases caused by nonsense mutations. Results of a preliminary trial of PTC124 in cystic fibrosis patients, published in the Aug. 30, 2008 issue of The Lancet, showed improvements in abnormalities caused by the mutation.
Now, in their study published in PNAS, researchers from the NIH Chemical Genomics Center (NCGC) present results that may have significant implications for the process used to identify PTC124, as well as other drug discovery efforts dependent upon firefly luciferase tests.
The NCGC's findings related to PTC124 arose from the center's efforts to optimize its own screening process by characterizing the types of chemical compounds that interfere with firefly luciferase. That work identified several families of chemical compounds that inhibit firefly luciferase.
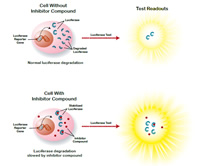
Most chemical compounds that inhibit firefly luciferase block or dim its signal. However, the NCGC researchers determined that some luciferase inhibitors — including PTC124 and related compounds belonging to the 3,5-diaryl-oxadiazole family — actually increase the signal's brightness by binding to the firefly enzyme in a way that slows its degradation within cells. In other words, the stabilized enzyme lights up even if the compound is inactive or only weakly active in the screening system.
"Our discovery is rather counter-intuitive. One wouldn't think that compounds that block firefly luciferase may actually increase its signal in some instances. But, in fact, that is exactly what our experiments found," said NCGC's Deputy Director James Inglese, Ph.D., who is senior author of the study. "This is a caveat that researchers now must consider when using firefly luciferase assays to screen for potential drugs."
Acting NIH Director Raynard S. Kington, M.D., Ph.D., said, "It is important to note that studies of PTC124 in patients and animal models of cystic fibrosis and Duchenne muscular dystrophy have produced encouraging early results, so it is unclear what impact these findings will have on the current clinical trials. Still, this new work will improve screening procedures and make our efforts to understand biology and develop therapeutics for many diseases more effective."
Dr. Inglese and his co-authors point out that researchers using firefly luciferase screening systems can double check their findings to ensure they are not getting false positive or exaggerated readouts in a number of ways. One approach is to retest compounds that appear to be active in firefly luciferase tests in a system that employs a different reporter. A reporter, in this case, is a lab term for any molecule used to signal a result. For example, the NCGC researchers found that PTC124 and its sister compounds do not inhibit a related reporter enzyme derived from Renilla reniformis, a type of soft coral commonly called a sea pansy. What's more, when the researchers used a sea pansy system to test PTC124's power to restore the ability of cells with nonsense mutations to produce full-length proteins, they detected no signs of activity.
"Our research emphasizes the need to understand interactions between reporter enzymes and chemical compounds, as well as to implement appropriate controls, when performing high-throughput screening," said the study's first author, Douglas S. Auld, Ph.D. If they have not already done so as part of their drug discovery process, Dr. Auld suggested that researchers using luciferase-based screens may want to consider conducting additional tests in screening systems that employ the sea pansy enzyme or other alternative reporters.
The NCGC was established as part of the NIH Roadmap for Medical Research and is part of the NIH's Molecular Libraries Probe Production Centers Network. The NCGC, which is administered by the National Human Genome Research Institute (NHGRI), is an ultra-high-throughput screening and chemistry center. The center's mission is to discover chemical probes that can serve as research tools to elucidate gene and cell functions or as starting points for the development of new therapeutics for rare and neglected diseases. NCGC researchers collaborate with more than 100 investigators from academic, non-profit and private sector laboratories throughout the world. To learn more about NCGC, go to www.ncgc.nih.gov.
Cystic fibrosis is an inherited disease that affects about 70,000 people worldwide. The disease causes the body to produce thick, sticky mucus that clogs the lungs, leading to life-threatening infections. It can also impair digestion by preventing vital enzymes from reaching the intestine. There currently is no cure for cystic fibrosis, but advances in supportive treatments now enable many people with the disease to live into their 30s and 40s or even beyond. While about 10 percent of cystic fibrosis patients have nonsense mutations, the percentage is much higher in certain populations, such as in Israel, where about 60 percent of patients have nonsense mutations. For more information on cystic fibrosis, go to www.genome.gov/10001213/learning-about-cystic-fibrosis/.
Duchenne muscular dystrophy is a progressive muscle-wasting disease that occurs mainly in boys. It affects about 1 in 3,500 males worldwide. There is no cure for the disease, and few people with Duchenne muscular dystrophy live beyond their 30s. For more information on Duchenne muscular dystrophy, go to www.genome.gov/19518854/learning-about-duchenne-muscular-dystrophy/.
To read the PNAS paper, go to Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression [pnas.org].
To download a high resolution graphic depicting how inhibitor compounds can slow the degradation of firefly luciferase, go to www.genome.gov/dmd/img.cfm?node=Photos/Graphics&id=85340.
NHGRI is one of 27 institutes and centers at the NIH, an agency of the Department of Health and Human Services. Additional information about NHGRI can be found at its Web site, www.genome.gov.
The National Institutes of Health (NIH) -"The Nation's Medical Research Agency" - includes 27 Institutes and Centers and is a component of the U. S. Department of Health and Human Services. It is the primary federal agency for conducting and supporting basic, clinical and translational medical research, and it investigates the causes, treatments and cures for both common and rare diseases. For more information about NIH and its programs, visit www.nih.gov.
Contact
Geoff Spencer, NHGRI
301-402-0911
spencerg@mail.nih.gov