Last updated: July 19, 2013

The 1000 Genomes Project more than doubles catalog of human genetic variation

National Human Genome Research Institute
www.genome.gov
The 1000 Genomes Project more than doubles catalog of human genetic variation
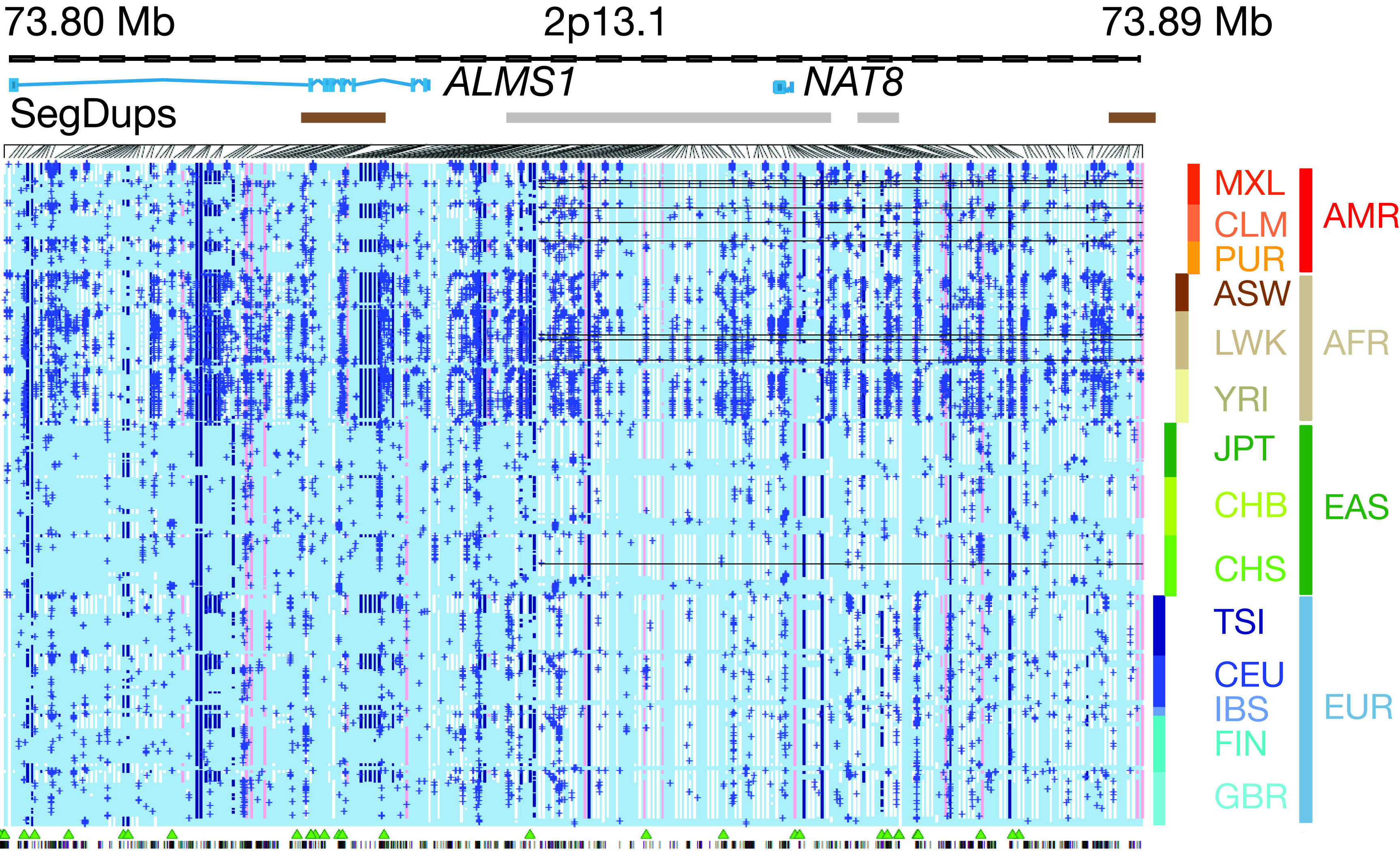 |
Genetic variation explains part of why people look different and vary in their risk for diseases. The goal of the 1000 Genomes Project is to identify and compile variants in the human genome that occur at a frequency of at least 1 in 50 people. Although most of these genetic variants cause little if any effect, some contribute to disease, and others are beneficial. An example of a beneficial difference is a rare genetic variant that blocks the human immunodeficiency virus from infecting white blood cells and, thus, protects people exposed to HIV who carry this variant. "
The 1000 Genomes Project is a large, international effort aiming to characterize human genetic variation, including people from many different populations," said Eric D. Green, M.D., Ph.D., NHGRI director. "The newly published findings provide deeper insights about the presence and pattern of variants in different people's genomes, which is critical information for studying the genomic basis of human disease."
The expanded catalog allows medical researchers to locate genetic differences contributing to rare and common diseases more precisely. Identifying the genetic underpinnings of disease will help lead to new diagnostic tests and, in some cases, treatments.
"I view this project as a Lewis and Clark expedition to the interior of the human genome," said Stephen Sherry, Ph.D., chief of the Reference Collections Section, Information Engineering Branch at the National Center for Biotechnology Information (NCBI), part of the National Library of Medicine. "We knew the outlines and contours (of the genome). Now, we're trying to document all the fine details such as the rivers and tributaries."
So far, project researchers have sequenced the genomes of 1,092 people from 14 populations in Europe, East Asia, sub-Saharan Africa and the Americas. Ultimately, they will study more than 2,500 individuals from 26 populations.
All of the participants consented to inclusion, in an open online database, of sequence data derived from their anonymous DNA samples. Each part of these genomes were read (or sequenced) an average of six times, which provides accurate information about common genetic variants but misses many rare variants.
To identify rare variants in the exome, the part of the genome that codes for proteins, the researchers sequenced the exons of 15,000 genes in each genome an average of 80 times. The study discovered 99.8 percent of exome variants with a frequency of at least 1 percent and 99.3 percent of variants elsewhere in the genome with a frequency of at least 1 percent.
Phase one of the 1000 Genomes Project, the subject of the Nature paper, has produced a massive amount of genomic data. Simply recording the raw information takes some 180 terabytes of hard-drive space, enough to fill more than 40,000 DVDs. All of the information is freely available on the Internet through public databases such as ones at the National Center for Biotechnology Information at the U.S. National Library of Medicine in Bethesda, Md., and the European Bioinformatics Institute in Hinxton, England. Data from the project have been available to researchers since 2008. The massive dataset became available in the cloud this year via Amazon Web Services (AWS). Cloud access enables users to analyze large amounts of the data much more quickly, as it eliminates the time-consuming download of data and because users can run their analyses over many servers at once. Researchers pay only for the additional AWS resources they need to further process or analyze the data.
"With this project, we have succeeded in making sure that information about our shared genetic heritage, and the common DNA variants we carry, are freely available for researchers to use to benefit patients around the world," said David Altshuler, M.D., Ph.D., an endocrinologist at Massachusetts General Hospital who directs the Broad Institute's Program in Medical and Population Genetics, and who co-leads the 1000 Genomes Project. "Moreover, the tools and methods that this project has helped foster are being used now in disease-oriented genetics research and will be used increasingly in clinical care."
All the genetic information for making an organism resides in the DNA, which is a set of long molecules made of units called bases. Each base is a chemical unit abbreviated A, C, G or T. For this paper, the researchers identified 38 million single-nucleotide polymorphisms, or SNPs (pronounced "snips"), which are DNA variants that occur when a particular basein the genome sequence differs among people.
These variants are the most common genetic differences among people. Each SNP is like a landmark, reflecting a specific position in the genome where the DNA spelling differs by one letter among people.
They also identified variants in the linear structure of the DNA, including 1.4 million short indels (insertions or deletions of DNA as small as a single base or as large as 50 bases) and 14,000 large deletions of DNA.
SNPs and structural variants can help explain an individual's susceptibility to disease, response to drugs, or reaction to environmental factors such as air pollution or stress. Other studies have found an elevated rate of indels in diseases such as autism and schizophrenia, although it's not yet clear how they affect those diseases.
"Project researchers discovered that each person carries a handful of rare variants that would currently be recognized as disease-causing and a few hundred more rare variants that are likely to have a detrimental effect on how genes work," said Gilean McVean, Ph.D., professor of statistical genetics at the University of Oxford in England and co-leader of the 1000 Genomes Project Analysis Group. "It's fortunate that most of us usually carry only one copy of these variants since two copies might lead to disease."
Another large NHGRI-funded effort, the ENCODE Project, recently published a series of papers showing that large parts of the human genome outside of protein-coding regions affect gene regulation. The patterns of variation in these regions that the 1000 Genomes Project found provide additional evidence about the functionality of these regions.
Data analysis is a vital part of the project, and about 260 analysts participated in analyzing the data reported in the recent Nature publication. They mapped and assembled the raw DNA sequence data relative to the reference human genome sequence. They then analyzed the aligned sequences to locate SNPs and structural variants. SNPs are relatively easy to find; structural variants (such as insertions, deletions and copy number differences) are much harder to find. A number of research groups are working to establish how to go from the raw sequence data to identifying structural variants.
Many research groups contributed to the generation of genome sequence data for this project, including NHGRI's large-scale sequencing centers: the Human Genome Sequencing Center at the Baylor College of Medicine, Houston; The Broad Institute of MIT and Harvard University in Cambridge, Mass., and The Genome Institute at the Washington University School of Medicine in St. Louis. Other groups included the Wellcome Trust Sanger Institute in Hinxton, England; BGI Shenzhen in Shenzhen, China; the Max Planck Institute for Molecular Genetics in Berlin; and Illumina, Inc., in San Diego.
The 1000 Genomes Project eliminates time-consuming steps for researchers trying to find genetic variants that affect a disease. Genome-wide association studies aim to find regions of the genome that contain DNA variants relevant to a disease. They use technologies that provide information about hundreds of thousands to a couple of million SNPs in each studied genome; they can combine these data with 1000 Genomes Project data on tens of millions of variants to find regions affecting the disease more precisely. The 1000 Genomes Project data then can be used to greatly enhance such studies by providing more detailed information about known variants. Instead of sequencing the genomes of all the people in a study - still an expensive prospect for thousands of people - researchers can use the 1000 Genomes Project data to find most of the variants in those regions that are associated with the disease.
"Once researchers find genes and variants of interest associated with disease by using the 1000 Genomes Project data, they have to return to basic biology to study them one at a time, to establish which genes and variants are causal for the disease and not just along for the ride," said Lisa D. Brooks, Ph.D., program director of the Genetic Variation Program in NHGRI's Division of Genome Sciences. "The 1000 Genomes Project data accelerate their ability to close in on those genes and variants."
Planning for the $120 million project began in 2007. In 2010, researchers published data on three pilot studies. The 2012 data set will be followed by the last addition to the catalog in 2013.
The 1000 Genomes Project data are available through:
- The 1000 Genomes website: www.1000genomes.org
- NCBI: ftp://ftp-trace.ncbi.nlm.nih.gov/1000genomes
- EBI: ftp://ftp.1000genomes.ebi.ac.uk
- Amazon: http://s3.amazonaws.com/1000genomes
NHGRI is one of the 27 institutes and centers at the National Institutes of Health, which is an agency of the U.S. Department of Health and Human Services. Additional information about NHGRI can be found at www.genome.gov.
About the National Institutes of Health (NIH): NIH, the nation's medical research agency, includes 27 Institutes and Centers and is a component of the U.S. Department of Health and Human Services. NIH is the primary federal agency conducting and supporting basic, clinical, and translational medical research, and is investigating the causes, treatments, and cures for both common and rare diseases. For more information about NIH and its programs, visit www.nih.gov.
Contact
Omar McCrimmon, NHGRI
301-402-0911
mccrimmono@mail.nih.gov