

Alexander Wilson, Ph.D.
Division of Intramural Research
B.A. Western Maryland College
Ph.D. Indiana University
-
Scientific Summary
The overarching goal of Dr. Wilson's research program has been the identification of genetic variants responsible for the variation in quantitative traits. The specific aims of this research program are: 1) to use statistical genetic analysis to identify genetic effects underlying quantitative traits and to identify statistical challenges that need to be addressed, 2) to develop new methods of genetic analysis for quantitative traits that address these challenges, 3) to use computer simulation to investigate the statistical properties of these methods, and 4) to apply insights gained from these simulations to ongoing collaborative studies. This work has spanned more than three decades, first at the Department of Biometry and Genetics at the Louisiana State University Medical Center and then the Genometrics Section of the Computational and Statistical Genomics Branch of the NHGRI Division of Intramural Research, NIH. This research has included applications projects and methods development in linkage analysis and tests of association with red-cell antigens, protein polymorphisms, STRPs, SNPs and next-generation sequence variants (SVs), in both family- and population-based samples.
Major substantive results include 1) the identification of genes for Mendelian syndromes (e.g., congenital cataracts, Cranio-Lenticulo-Sutural-Dysplasia), 2) the identification of polymorphisms responsible for variation in quantitative traits (e.g., dopamine-beta-hydroxylase activity, citalopram response in depressed individuals, and platelet aggregation), and 3) the identification of candidate regions with linkage and association in complex disorders (e.g., traits related to hypertension and cardiovascular disease, depression and alcoholism, familial idiopathic scoliosis and kyphoscoliosis, and craniosynostosis).
Methodological work includes: 1) advances in non-parametric linkage analysis, 2) stepwise regression of identified variants in quantitative results, 3) regional inference with moving averages of p-values, 4) the use of derived composite biallelic loci, 5) testing associations in parent-offspring trios with a regression of offspring on mid-parent (ROMP) based approach, and 6) the use of hot-spot based delimiters to divide the genome into independent segments in a linear regression format for family- and population-based tests of association for sequence variants (tiled regression). In addition, the large and small sample statistical properties of these tests have been investigated with computer simulation studies to ensure that the tests are statistically valid and have reasonable power and type I error rates. Software packages developed include 1) the Genometric Simulation Analysis Package (GASP), 2) the implementation of the Regression of Offspring on Mid-Parent (ROMP, ROOP and ROMPrev) and 3) the implementation of the tiled regression approach, the Tiled Regression Analysis Package (TRAP). These packages are available on the NHGRI website.
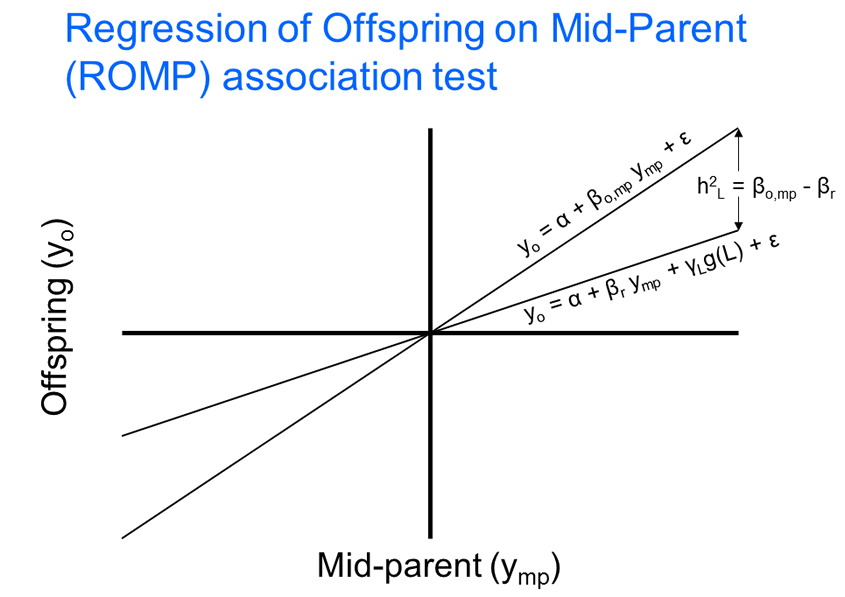
During the last several years, the density of genetic variants has increased dramatically and the section's research has become particularly focused on approaches that are robust with respect to the non-independence between markers and correlations between family members within samples, and on the identification of non-coding regulatory elements. Recent methodological work has focused on the development of two new methods for quantitative traits: the regression of offspring on mid-parent (ROMP), and tiled regression. Both are tests of association in a linear regression framework that can be applied to family data and have been designed for data with very large numbers of genetic markers (millions), e.g. high density SNP panels, and/or large-scale sequencing producing large numbers of rare sequence variants. ROMP is designed to minimize the amount of genotyping and/or sequencing required for a test of association in a parent-offspring trio or nuclear family, by requiring phenotyping data on the parents and offspring, but requiring genotyping or sequencing on only the offspring. Tiled regression is a method that determines the set of independent sequence variants across the entire genome that best predict a given phenotype, against the background of all the variants in the genome.
Regression methods are used to identify independent variants (both coding and non-coding) in predefined independent tiles, that are defined by hotspot blocks, or other positional or functional regions. Higher level regression is then used to determine independent variants over chromosomes and the entire genome. At the Genetic Analysis Workshop 17 (GAW 17), in 2010, it was discovered that there was a substantial inflation of type I error when GWAS methods were used to analyze rare sequence variants, most likely due to the presence of gametic disequilibrium (or inter-LD). The tiled regression method was one of the only methods that allowed for both intra- and inter-LD correlations and it did not exhibit inflation of type I error rates that were present in virtually all of the other methods considered. Before this workshop, it was generally assumed that adjustments only had to be made for correlations within LD blocks (intra-LD correlations); but based on the GAW 17 findings, this is clearly not the case.
Future plans include extensions of TRAP to qualitative traits, and the inclusion of tiled regression into the regression of offspring on mid-parent (ROMP) regression framework. The tiled regression approach, using both hotspot and functional criteria to define tiles, is currently being used in all of our ongoing and future collaborative studies. These projects include 1) analysis of all ClinSeq traits and sequence data (Les Biesecker et al.), 2) analysis of the Trinity Irish metabolite data (Lawrence Brody), 3) functional studies in zebrafish based on our kyphoscoliosis IRX results (Nancy Miller), 4) whole exome sequencing and analysis of a large family with metopic craniosynostosis (Simeon Boyadjiev), and 5) targeted or whole exome sequencing and analysis of about 70 families with familial idiopathic scoliosis (Nancy Miller).
Last updated: June 21, 2022