

Michael R. Erdos, Ph.D.
Molecular Genetics Section
B.S., The George Washington University
Ph.D., University of Groningen
Scientific Summary
The application of genetic, genomic and functional biology approaches are valuable and increasingly powerful ways to provide understanding of both rare and complex diseases and of the potential for therapeutic development.
Hutchinson-Gilford Progeria Syndrome
In the ultra-rare premature aging disorder HGPS, Dr. Erdos and colleagues' lab discovered the dominant negative cryptic splice mutation, c.1824 C/T, in the LMNA gene generates the mutant progerin protein responsible for HGPS and causes a plethora of phenotypic consequences, including death from heart attack of stroke by the early teens. But there is also much hope. In fact, HGPS can be seen as a textbook demonstration of the evolution of therapeutic advances for a rare disorder. Normally, the LMNA gene precursor protein, prelamin A, is farnesylated prior to incorporation into the nuclear membrane, then cleaved by an endoprotease to release mature lamin A that is incorporated in the nuclear lamina matrix. The deletion of the 50 amino acids in the mutant progerin protein created by the cryptic splice mutation results in the permanent farnesylation of mature lamin A causing disruption of nuclear organization and chromatin structure. The cellular consequence is premature senescence, which contributes to the accelerated aging phenotype.
Because this mutation directs a dominant negative effect, it facilitated the generation of a humanized transgenic mouse model by inserting a human genomic DNA segment containing the LMNA gene engineered with the c.1824C/T mutation. In the heterozygous state the mouse model embodies the characteristic vascular defects found in HGPS patients, including age-related loss of vascular smooth muscle cells (VSMCs) and stiffening of arterial walls. In the homozygous state the mouse presents with many additional phenotypes characteristics of HGPS including retarded growth, lipodystrophy, joint contractures and premature death at a median of 210 days.
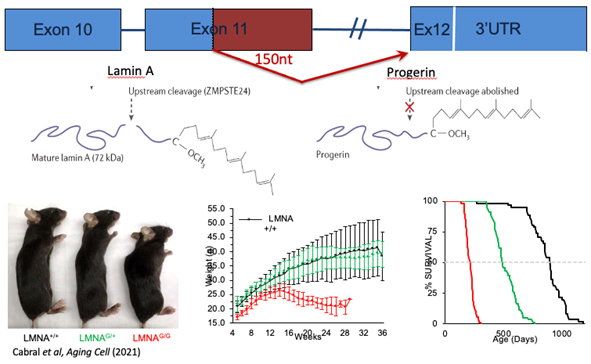
The HGPS mouse is a highly useful model for preclinical investigation of potential therapeutics, particularly for genetic therapies, because the effects are derived from the expression of progerin from human LMNA DNA sequence.
In the realm of small molecule therapeutics, farnesyl transferase inhibitors (FTIs) target the specific defect in progerin that generates the dominant negative cytotoxic effect. Dr. Erdos and colleagues reasoned that blocking the permanent addition of the farnesyl hydrophobic tail would reduce the deleterious effects of the mutant progerin protein. Treating the mouse model with FTIs was successful at reducing the cardiovascular defects in young mice. In addition, it improved the cardiovascular histopathology when administered to older mice. This work supported the first clinical trial application for the use of FTIs to treat children with HGPS, conducted by collaborators at Boston Children’s Hospital. That trial showed improvement in arterial stiffness, and ultimately a modest extension in lifespan. FDA approved lonafarnib (a farnesyltransferase inhibitor) for HGPS in 2021.
In a complementary approach, the hypothesis that rapamycin analogues, capable of inhibiting the mTOR pathway, would reduce the deleterious effects of progerin by stimulating cellular autophagy was tested in the mouse model. Treatment of the HGPS mouse model with rapamycin resulted in improved cardiovascular histopathology and moderately increased survival. These results were validated by crossing the mouse model with an mTOR hypomorphic genetic mouse model provided by Dr. Toren Finkel, exhibiting reduced mTOR expression achieved similar benefits. A trial of the rapalog everolimus, combined with lonafarnib, is currently underway.
In a more direct effort to target the genetic cause of HGPS, Dr. Erdos and colleagues are employing antisense oligomer splice inhibition therapeutics to reduce progerin production at the mRNA level by blocking the mutant splice site. Treatment of the homozygous mouse model with the c.1824 C/T mutation specific peptide-conjugated phosphorodiamidate morpholino-oligomer (PPMO), SRP-2001, improved the cardiovascular state and increased the lifespan of the homozygous mouse model by 62%. Dr. Erdos and colleagues are now supporting the Progeria Research Foundation application with the FDA for treatment of HGPS children with antisense oligomers.
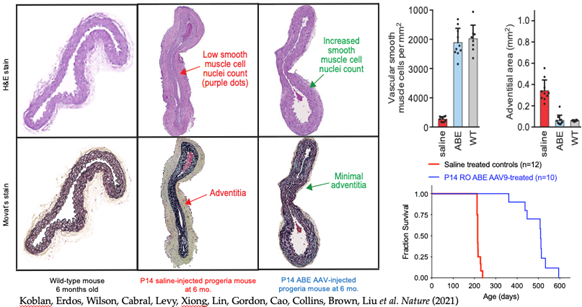
Antisense oligomer therapeutics are safe and more effective than the current small molecule therapeutics (FTIs and rapamycin), but the treatment regimen requires frequent injections for the lifetime of the patient. More recently, the laboratory of Dr. David Liu at the Broad Institute has developed the CRISPR-mediated Adenine-deaminase DNA Base Editor (ABE) packaged in adeno-associated virus (AAV9) customized for the c.1824C/T mutation. This requires only a single intravenous administration, a perfect candidate for therapeutic development for HGPS. In collaboration with the Liu laboratory, Dr. Erdos and colleagues have shown that the AAV9-ABE-c.1824 C/T DNA base editors are capable of systemic delivery in mice and can achieve 8-30% mutation correction in critical HGPS tissues. Somatic selection further provides an advantage – as the uncorrected cells senesce, the corrected cells seem to take their place: six months post treatment the large arteries are fully populated with VSMCs negative for progerin staining. In a longevity trial the treated mice achieve a 2.4-fold increase in lifespan. Dr. Erdos is now collaborating on the development of ABEs for FDA application by Beam Therapeutics and the Progeria Research Foundation.
Type 2 Diabetes
Type 2 diabetes is a complex multi-organ system disorder characterized by dysregulation of glucose and fatty acid sensing and synthesis in the liver, insulin resistance in the adipose and muscle, and ultimately failure to compensate for high blood sugar by insulin secretion from the pancreatic islets. Complex diseases like T2D present a formidable challenge for translational research into potential therapeutics. Although there is an established genetic component underlying T2D susceptibility, environmental conditions such as diet and physical activity are a significant contribution to the prevalence of T2D in populations. Based on work by Dr. Erdos and colleagues' group and others, there are over 400 genetic variants significantly associated with T2D. Over 90% of these fall in non-coding regions of the genome, making it difficult to determine what gene has its expression influenced by the variant. Dr. Erdos’s T2D research currently focuses on the development of functional genomics analyses of model systems to determine the causal effect of T2D associated variants, identify the target genes associated with the variants and define their role in the etiology of T2D.
Dr. Erdos is the lead scientist in the FUSION tissue biopsy project which aims to investigate tissue specific effects of T2D associated variants on the critical tissues involved in T2D. The FUSION study collected plasma, muscle and adipose from normal glucose tolerant (NGT) subjects, impaired glucose tolerant (IGT) and newly diagnosed T2D subjects not on medication from the Finnish population in order to perform transcriptomics, epigenomics and gene-environment interaction analyses correlated with T2D associated variants. In addition, skin biopsies were obtained from each subject for fibroblast cell culture and induced pluripotent stem cell (iPSC) generation, with the goal to differentiate the iPSCs into tissues for functional investigation of T2D associated variants. To complement the tissues accessible from FUSION subjects, Dr. Erdos has been collecting pancreatic islets and liver tissue from cadaveric tissue distribution organizations in order to enable complementary T2D variant analyses in organ systems important in T2D but not attainable from the FUSION subjects. The aim of these studies is to compile the knowledge gained from studying these multi-organ genetic associations to classify subtypes of T2D for the identification of potential therapies based on the class of diabetes presented.
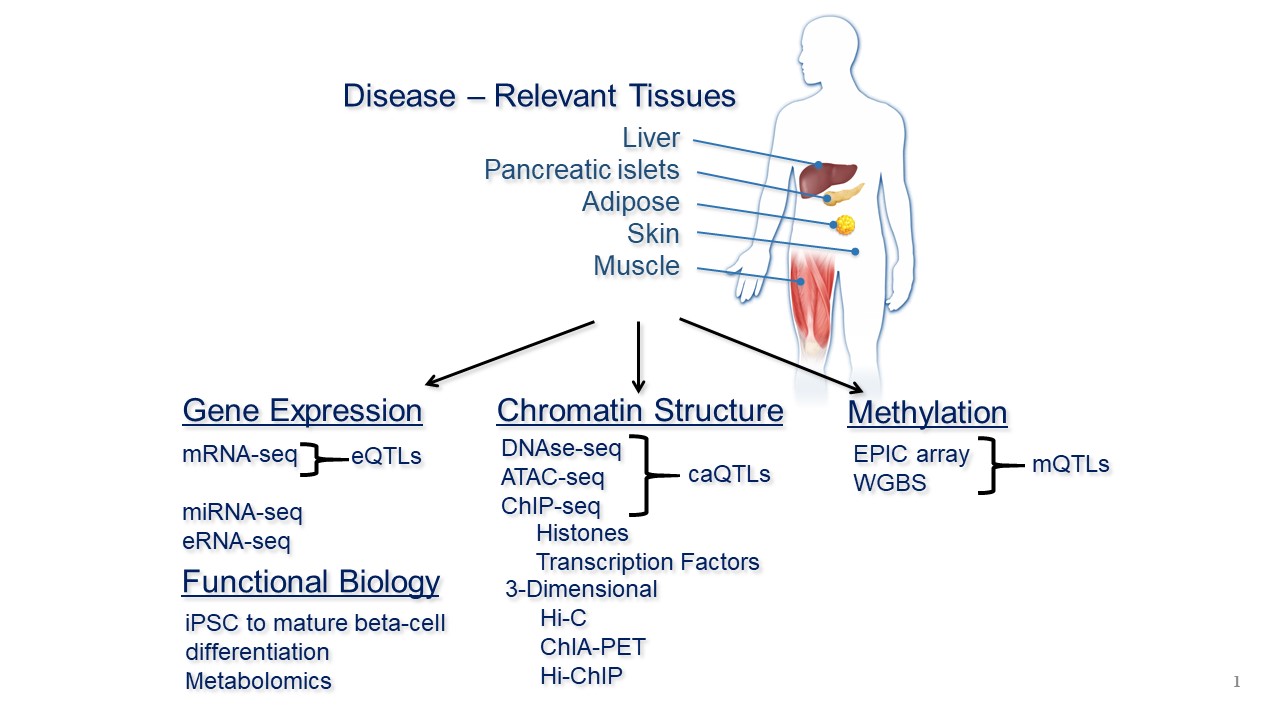
The primary pancreatic islet model system Dr. Erdos is developing is the automated differentiation of induced pluripotent stem cells (iPSCs) into mature pancreatic beta cells, carried out in collaboration with the automation experts at the New York Stem Cell Foundation. Dr. Erdos selected 52 individuals from the FUSION Tissue Biopsy Study, either normal glucose tolerant, impaired glucose tolerant or newly diagnosed untreated T2D subjects, and is examining the differentiation activity of these lines using single-cell multi-omic integrated analysis of chromatin structure and gene expression with glucose stimulated insulin secretion (GSIS) functional analysis. The cell state and functional consequence are integrated with genotype and polygenic risk to investigate the functional impact of T2D-associated variants and identify potential targets for focused therapeutic development. For known causal variants Dr. Erdos and colleagues are also able to employ CRISPR-based DNA editing to create isogenic iPSC lines for allelic evaluation using this automated system. In addition, Dr. Erdos and colleagues are employing this system in CRISPRi -genome wide gene inhibition experiments to identify critical genes required for development and maturity of pancreatic beta cells and functional response to glucose and insulin secretagogues. Their ultimate goal is to determine the most promising target genes in these T2D functional networks for the development of precision therapeutics.
Molecular Genetics Section Staff

- Predoctoral IRTA Fellow
- Molecular Genetics Section

- Postdoctoral Fellow
- Molecular Genetics Section

- Bioinformatics Scientist
- Molecular Genetics Section

- Guest Researcher
- Molecular Genetics Section

- Jr. Scientific Data Specialist
- Molecular Genetics Section
- Postbaccalaureate IRTA Fellow
- Molecular Genetics Section

- Postbaccalaureate IRTA Fellow
- Molecular Genetics Section

- Postbaccalaureate IRTA Fellow
- Molecular Genetics Section
- Special Volunteer
- Molecular Genetics Section

- Special Volunteer
- Molecular Genetics Section
Last updated: November 7, 2024