

Settara Chandrasekharappa, Ph.D.
Cancer Genomics Unit
Genomics Core
Cancer Genomics Unit
B.Sc. (Hons) Bangalore University, 1972
M.Sc. Bangalore University, 1974
Ph.D. Indian Institute of Science, 1983
-
Scientific Summary
The Cancer Genomics Unit (CGU) focuses on adopting genomic technologies for identification of disease genes and the underlying molecular mechanisms leading to pathogenesis. Between 1991 and 1994, as a member of the collaborative group in search of the gene responsible for early onset breast cancer (BRCA1), CGU developed resources and aided in the construction of genetic, physical and transcript maps of the locus on 17q. The advent of large insert genomic libraries allowed the CGU to participate in physical mapping efforts of disease gene regions and positional cloning efforts of genes involved in Menkes disease, Parkinson's disease, and Bardet-Biedl syndrome 1. CGU developed and employed BAC arrays for exploring the genomic changes in chromosome 8 in breast and colon cancer.
In 1997, Dr. Chandrasekharappa's lab was successful in the positional cloning of the gene responsible for Alagille syndrome (AGS), a congenital disorder characterized by paucity of bile ducts in the liver and abnormalities of the heart, eyes, vertebrae and face. Mutations in the JAG1 gene, which encodes a ligand (JAGGED 1) for the Notch transmembrane receptor, was found to be responsible for AGS. The Notch signaling pathway, originally discovered in fruit flies, is important in determining early cell-fate decisions. The Chandrasekharappa lab participated in positional cloning of mib, a gene in the Notch pathway in zebrafish. Mutations in mib result in increased neural progenitor differentiation to neurons during embryonic development, leading to the zebrafish mind bomb phenotype.
In a collaborative effort in 1997, Dr. Chandrasekharappa's lab was also successful in positional cloning of the gene responsible for multiple endocrine neoplasia type 1 (MEN1), a condition characterized by tumors primarily of the parathyroid, pituitary and endocrine pancreas, and caused by mutations in a novel tumor suppressor gene, MEN1. This research effort in the Chandrasekharappa lab centered on understanding the biological functions of the MEN1-encoded protein, menin. The lab further investigated the expression of menin in mouse, zebrafish and drosophila development, and they also investigated menin's interaction with several different proteins, including the transcription factor JunD, and its requirement in endodermal differentiation. The lab made several original contributions to the fields of endocrine tumorigenesis research. CGU concluded studies on MEN1 in 2009 with a report that menin is required for in vitrohematopoietic differentiation of mouse embryonic stem cells and this role of menin is mediated by its ability to regulate Hoxa9 expression.
In 2009, CGU switched its focus to Fanconi anemia (FA), a rare recessive disorder characterized by debilitating congenital abnormalities, life-threatening bone marrow failure, and a predisposition to myeloid, head and neck and other malignancies. Because FA is caused by mutations in at least 16 genes in DNA repair pathways, nearly half of FA patients lack proper molecular diagnosis. Understanding the underlying molecular defect is critical for complete diagnosis and subsequent clinical management. The Chandrasekharappa lab designed a high-density array comparative genomic hybridization (arrayCGH) platform to scan all 16 FA genes for deletions and duplications, and effectively utilized next-generation sequencing strategies for mutation screening. After finding exon skipping arising from a deep intronic mutation in one patient, and from a synonymous change in a second patient, the lab began to include RNAseq along with DNAseq as a part of FA mutation screening efforts. These findings helped develop an efficient strategy for identifying mutations in nearly 300 FA families. Pinpointing specific mutations and their pathogenicity will lead to a better understanding of the phenotypic heterogeneity in this disorder.
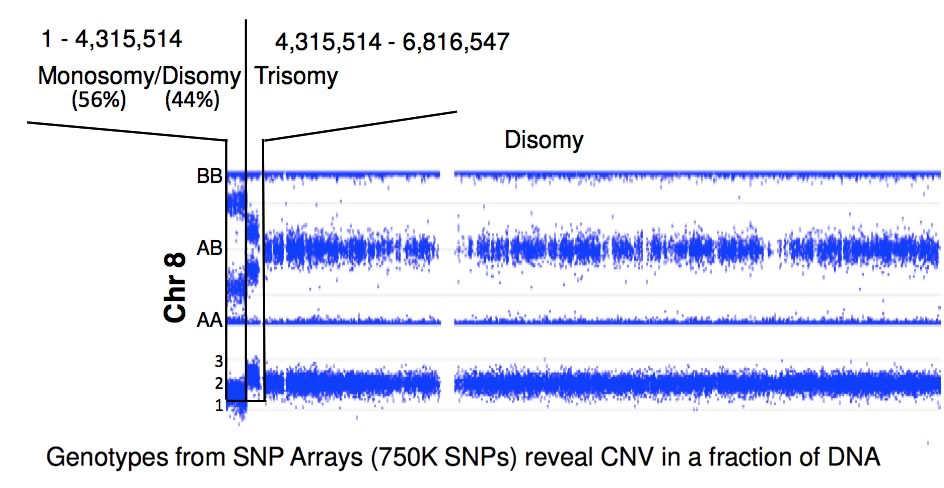
Because FA patients display genomics instability, the Chandrasekharappa lab employed high-density SNP arrays to detect the presence of de novo deletions or duplications, loss, gain, loss of heterozygosity or uniparental disomy. These changes will provide clues to FA's considerable phenotypic heterogeneity. Nearly a quarter of FA patients display somatic mosaicism, a phenomenon which occurs due to a suppressor mutation complementing the initial FA-causing mutation. The CGU evaluates the molecular changes leading to mosaicism and explores the mechanisms of their occurance. By correlating the presence of mosaicism with phenotypic variables, the CGU will assess the impact of mosaicism on disease progression and study its relationship to treatment.
Increased incidence of head and neck cancer in patients diagnosed with FA is a major concern, as FA patients cannot tolerate conventional cancer therapies. It is possible that head and neck cancer could be the first manifestation of FA disease in some patients. In order to explore the extent of undiagnosed FA in head and neck cancer patients, and identify the prevalence of FA mutations in this population, the CGU is screening for FA gene mutations in young adults with head and neck cancer. Understanding these genomic changes and their relationship to phenotypic heterogeneity may open up opportunities for better treatment options.
Last updated: December 6, 2021