
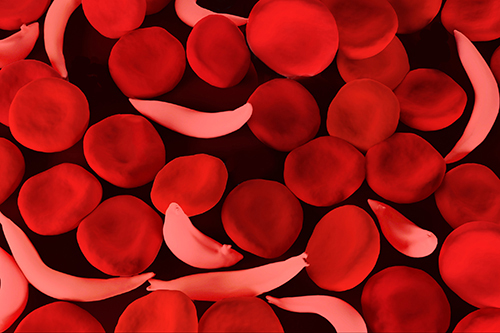
Sickle cell disease community hopeful genome editing will rechart course of disease
About Sickle Cell Disease
Sickle cell disease is a group of inherited red blood cell disorders.
What do we know about heredity and sickle cell disease?
Sickle cell disease is the most common inherited blood disorder in the United States. Approximately 100,000 Americans have the disease.
In the United States, sickle cell disease is most prevalent among African Americans. About one in 12 African Americans and about one in 100 Hispanic Americans carry the sickle cell trait, which means they are carriers of the disease.
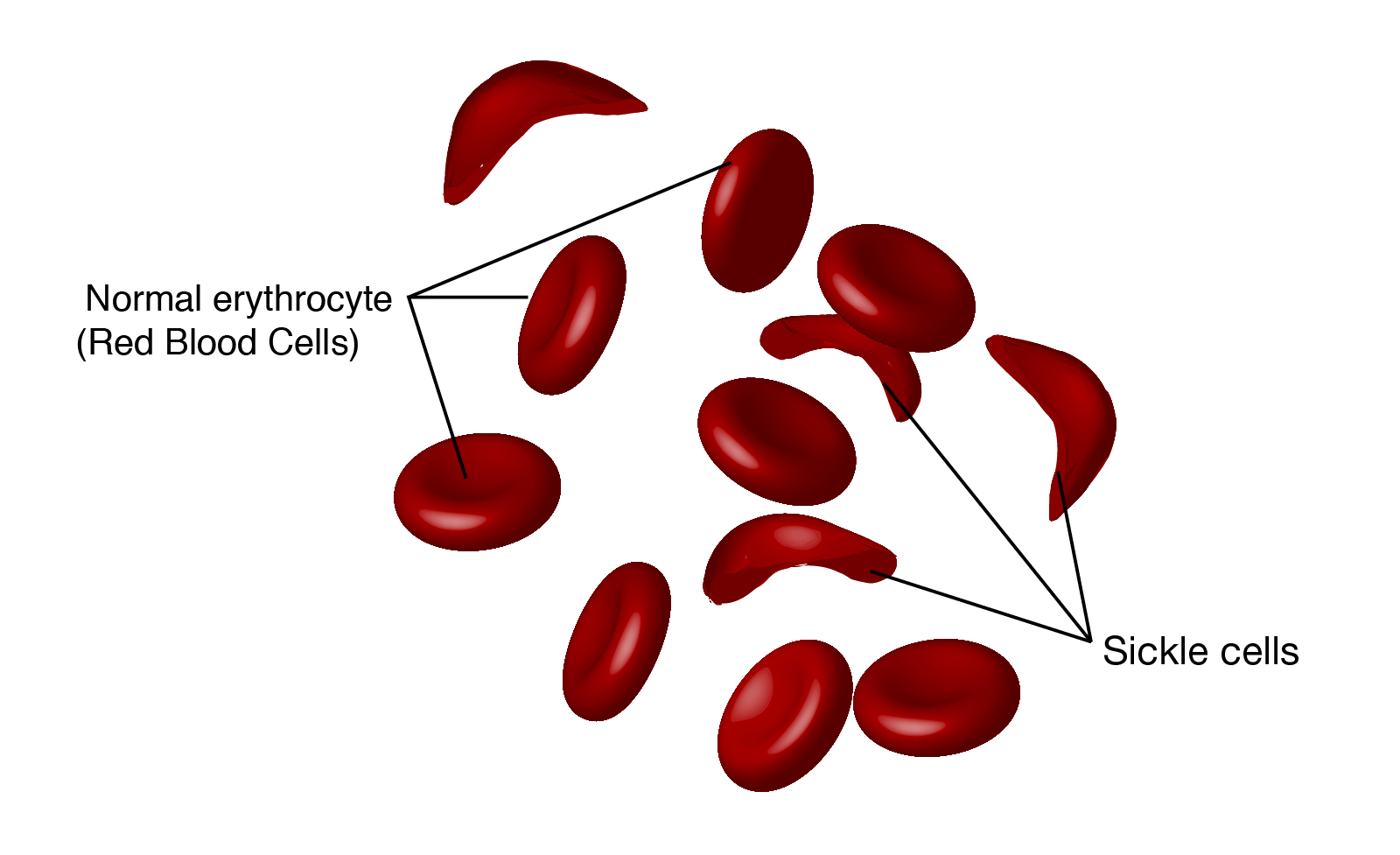
Sickle cell disease is caused by a mutation in the hemoglobin-Beta gene found on chromosome 11. Hemoglobin transports oxygen from the lungs to other parts of the body. Red blood cells with normal hemoglobin (hemoglobin-A) are smooth and round and glide through blood vessels.
In people with sickle cell disease, abnormal hemoglobin molecules - hemoglobin S - stick to one another and form long, rod-like structures. These structures cause red blood cells to become stiff, assuming a sickle shape. Their shape causes these red blood cells to pile up, causing blockages and damaging vital organs and tissue.
Sickle cells are destroyed rapidly in the bodies of people with the disease, causing anemia. This anemia is what gives the disease its commonly known name - sickle cell anemia.
The sickle cells also block the flow of blood through vessels, resulting in lung tissue damage that causes acute chest syndrome, pain episodes, stroke and priapism (painful, prolonged erection). It also causes damage to the spleen, kidneys and liver. The damage to the spleen makes patients - especially young children - easily overwhelmed by bacterial infections.
A baby born with sickle cell disease inherits a gene for the disorder from both parents. When both parents have the genetic defect, there's a 25 percent chance that each child will be born with sickle cell disease.
If a child inherits only one copy of the defective gene (from either parent), there is a 50 percent chance that the child will carry the sickle cell trait. People who only carry the sickle cell trait typically don't get the disease, but can pass the defective gene on to their children.
New Treatments Prolong Life:
Until recently, people with sickle cell disease were not expected to survive childhood. But today, due to preventive drug treatment, improved medical care and aggressive research, half of sickle cell patients live beyond 50 years.
Treatments for sickle cell include antibiotics, pain management and blood transfusions. A new drug treatment, hydroxyurea, which is an anti-tumor drug, appears to stimulate the production of fetal hemoglobin, a type of hemoglobin usually found only in newborns. Fetal hemoglobin helps prevent the "sickling" of red blood cells. Patients treated with hydroxyurea also have fewer attacks of acute chest syndrome and need fewer blood transfusions.
Bone Marrow Transplantation: The Only Cure:
Currently the only cure for sickle cell disease is bone marrow transplantation. In this procedure a sick patient is transplanted with bone marrow from healthy, genetically compatible sibling donors. However only about 18 percent of children with sickle cell disease have a healthy, matched sibling donor. Bone marrow transplantation is a risky procedure with many complications.
Gene Therapy Offers Promise of a Cure:
Researchers are experimenting with attempts to cure sickle cell disease by correcting the defective gene and inserting it into the bone marrow of those with sickle cell to stimulate production of normal hemoglobin. Recent experiments show promise.
Researchers used bioengineering to create mice with a human gene that produces the defective hemoglobin causing sickle cell disease. Bone marrow containing the defective hemoglobin gene was removed from the mice and genetically "corrected" by the addition of the anti-sickling human beta-hemoglobin gene. The corrected marrow was then transplanted into other mice with sickle cell disease. The genetically corrected mice began producing high levels of normal red blood cells and showed a dramatic reduction in sickled cells. Scientists are hopeful that the techniques can be applied to human gene transplantation using autologous transplantation, in which some of the patient's own bone marrow cells would be removed and genetically corrected.
-
What do we know about heredity and sickle cell disease?
Sickle cell disease is the most common inherited blood disorder in the United States. Approximately 100,000 Americans have the disease.
In the United States, sickle cell disease is most prevalent among African Americans. About one in 12 African Americans and about one in 100 Hispanic Americans carry the sickle cell trait, which means they are carriers of the disease.
Sickle cell disease is caused by a mutation in the hemoglobin-Beta gene found on chromosome 11. Hemoglobin transports oxygen from the lungs to other parts of the body. Red blood cells with normal hemoglobin (hemoglobin-A) are smooth and round and glide through blood vessels.
In people with sickle cell disease, abnormal hemoglobin molecules - hemoglobin S - stick to one another and form long, rod-like structures. These structures cause red blood cells to become stiff, assuming a sickle shape. Their shape causes these red blood cells to pile up, causing blockages and damaging vital organs and tissue.
Sickle cells are destroyed rapidly in the bodies of people with the disease, causing anemia. This anemia is what gives the disease its commonly known name - sickle cell anemia.
The sickle cells also block the flow of blood through vessels, resulting in lung tissue damage that causes acute chest syndrome, pain episodes, stroke and priapism (painful, prolonged erection). It also causes damage to the spleen, kidneys and liver. The damage to the spleen makes patients - especially young children - easily overwhelmed by bacterial infections.
A baby born with sickle cell disease inherits a gene for the disorder from both parents. When both parents have the genetic defect, there's a 25 percent chance that each child will be born with sickle cell disease.
If a child inherits only one copy of the defective gene (from either parent), there is a 50 percent chance that the child will carry the sickle cell trait. People who only carry the sickle cell trait typically don't get the disease, but can pass the defective gene on to their children.
New Treatments Prolong Life:
Until recently, people with sickle cell disease were not expected to survive childhood. But today, due to preventive drug treatment, improved medical care and aggressive research, half of sickle cell patients live beyond 50 years.
Treatments for sickle cell include antibiotics, pain management and blood transfusions. A new drug treatment, hydroxyurea, which is an anti-tumor drug, appears to stimulate the production of fetal hemoglobin, a type of hemoglobin usually found only in newborns. Fetal hemoglobin helps prevent the "sickling" of red blood cells. Patients treated with hydroxyurea also have fewer attacks of acute chest syndrome and need fewer blood transfusions.
Bone Marrow Transplantation: The Only Cure:
Currently the only cure for sickle cell disease is bone marrow transplantation. In this procedure a sick patient is transplanted with bone marrow from healthy, genetically compatible sibling donors. However only about 18 percent of children with sickle cell disease have a healthy, matched sibling donor. Bone marrow transplantation is a risky procedure with many complications.
Gene Therapy Offers Promise of a Cure:
Researchers are experimenting with attempts to cure sickle cell disease by correcting the defective gene and inserting it into the bone marrow of those with sickle cell to stimulate production of normal hemoglobin. Recent experiments show promise.
Researchers used bioengineering to create mice with a human gene that produces the defective hemoglobin causing sickle cell disease. Bone marrow containing the defective hemoglobin gene was removed from the mice and genetically "corrected" by the addition of the anti-sickling human beta-hemoglobin gene. The corrected marrow was then transplanted into other mice with sickle cell disease. The genetically corrected mice began producing high levels of normal red blood cells and showed a dramatic reduction in sickled cells. Scientists are hopeful that the techniques can be applied to human gene transplantation using autologous transplantation, in which some of the patient's own bone marrow cells would be removed and genetically corrected.
Is there a test for sickle cell disease?
Doctors diagnose sickle cell through a blood test that checks for hemoglobin S - the defective form of hemoglobin. To confirm the diagnosis, a sample of blood is examined under a microscope to check for large numbers of sickled red blood cells - the hallmark trait of the disease.
In more than 40 states, testing for the defective sickle cell gene is routinely performed on newborns.
Sickle cell disease can also be detected in an unborn baby. Amniocentesis, a procedure in which a needle is used to take fluid from around the baby for testing, can show whether the fetus has sickle cell disease or carries the sickle cell gene. If the test shows that the child will have sickle cell disease, some parents may choose not to continue the pregnancy. Genetic counselors can help parents make these difficult decisions.
A new technique used in conjunction with in vitro fertilization, called pre-implantation genetic diagnosis (PGD), enables parents who carry the sickle cell trait to test embryos for the defective gene before implantation, and to choose to implant only those embryos free of the sickle cell gene.
Additional Resources for Sickle Cell Disease Information
-
Is there a test for sickle cell disease?
Doctors diagnose sickle cell through a blood test that checks for hemoglobin S - the defective form of hemoglobin. To confirm the diagnosis, a sample of blood is examined under a microscope to check for large numbers of sickled red blood cells - the hallmark trait of the disease.
In more than 40 states, testing for the defective sickle cell gene is routinely performed on newborns.
Sickle cell disease can also be detected in an unborn baby. Amniocentesis, a procedure in which a needle is used to take fluid from around the baby for testing, can show whether the fetus has sickle cell disease or carries the sickle cell gene. If the test shows that the child will have sickle cell disease, some parents may choose not to continue the pregnancy. Genetic counselors can help parents make these difficult decisions.
A new technique used in conjunction with in vitro fertilization, called pre-implantation genetic diagnosis (PGD), enables parents who carry the sickle cell trait to test embryos for the defective gene before implantation, and to choose to implant only those embryos free of the sickle cell gene.
Additional Resources for Sickle Cell Disease Information
Related Content
Last updated: May 26, 2020