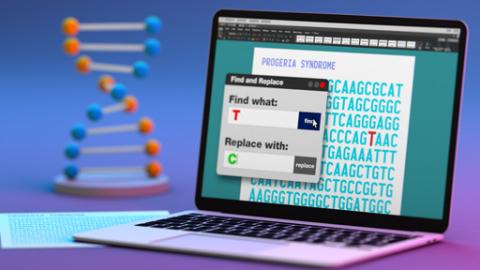
Research News and Events Sort by Most RecentHeadline (A-Z) Browse by Month JAN FEB MAR APR MAY JUN JUL AUG SEP OCT NOV DEC JUL 2026 Event NHGRI Division of Intramural Research Seminar Series Read more Event Jeffrey M. Trent Lectureship in Cancer Research Series Read more Event Social and Behavioral Research Branch Seminar Series Read more Event Scientific Updates on Organic Acidemias and Homocystinurias Read more News Release DNA-editing method shows promise to treat mouse model of progeria Read more News Release NIH and Alaska Native leaders identify how to achieve socially responsible genomics research Read more News Release Alexander Wilson, a statistical geneticist with poetic sensibilities, retires with a guitar in hand Read more News Release A new tool to prevent the spread of hospital-acquired infections in the era of COVID-19 Read more News Release Decades of NIH research help lead to first FDA-approved treatment for progeria Read more View More Last updated: March 21, 2019