
GNE Myopathy
The Natural History Study of Patients with GNE Myopathy collects genetic and medical information from people with GNE myopathy. Patients are followed over time to understand the symptoms and clinical course of GNE myopathy. This information is essential to prepare clinical treatment trials.
What is GNE Myopathy?
GNE Myopathy is a rare (autosomal recessive) genetic disorder that causes progressive skeletal muscle atrophy and weakness. Previous names include hereditary inclusion body myopathy (HIBM), inclusion body myopathy type 2 (IBM2) or Nonaka myopathy.
Symptoms of the disease usually appear between 20 and 40 years of age and include foot drop and difficulty walking. The disease slowly affects other muscles of the arms and legs. Patients typically start using a wheelchair one or two decades later and eventually some patients may need assistance with activities of daily living.
GNE Myopathy results from variants in a gene called GNE, which is responsible for a step in the production of a sugar called sialic acid. The diagnosis may be suggested by the presence of red-rimmed vacuoles (inclusions) in muscle biopsy tissue. Identification of GNE gene disease-causing variants by DNA sequencing can confirm the diagnosis.
GNE Myopathy patients may go undiagnosed or misdiagnosed with a different disease for many years. There are ~2,000 patients known worldwide and ~200 in the United States. However, we estimate (based on examining DNA sequencing databases) that there are at least 40,000 GNE Myopathy patients worldwide, including ~13,000 in Asia (~750 in Japan), ~4,000 in Europe and ~3,000 in North America, meaning that many patients are undiagnosed.
No therapies are currently approved for GNE Myopathy. Studies on mouse models of GNE Myopathy have provided promising data and several therapeutic trials are currently underway. NIH investigators are developing ManNAc as a therapy for GNE Myopathy.
-
What is GNE Myopathy?
GNE Myopathy is a rare (autosomal recessive) genetic disorder that causes progressive skeletal muscle atrophy and weakness. Previous names include hereditary inclusion body myopathy (HIBM), inclusion body myopathy type 2 (IBM2) or Nonaka myopathy.
Symptoms of the disease usually appear between 20 and 40 years of age and include foot drop and difficulty walking. The disease slowly affects other muscles of the arms and legs. Patients typically start using a wheelchair one or two decades later and eventually some patients may need assistance with activities of daily living.
GNE Myopathy results from variants in a gene called GNE, which is responsible for a step in the production of a sugar called sialic acid. The diagnosis may be suggested by the presence of red-rimmed vacuoles (inclusions) in muscle biopsy tissue. Identification of GNE gene disease-causing variants by DNA sequencing can confirm the diagnosis.
GNE Myopathy patients may go undiagnosed or misdiagnosed with a different disease for many years. There are ~2,000 patients known worldwide and ~200 in the United States. However, we estimate (based on examining DNA sequencing databases) that there are at least 40,000 GNE Myopathy patients worldwide, including ~13,000 in Asia (~750 in Japan), ~4,000 in Europe and ~3,000 in North America, meaning that many patients are undiagnosed.
No therapies are currently approved for GNE Myopathy. Studies on mouse models of GNE Myopathy have provided promising data and several therapeutic trials are currently underway. NIH investigators are developing ManNAc as a therapy for GNE Myopathy.
What is Involved?
This study collects genetic and medical information from people with GNE Myopathy and other GNE-related disorders. Patients are followed over time to understand the symptoms and clinical course of GNE Myopathy and related disorders. This information is essential to prepare clinical treatment trials.
To be eligible to participate, individuals must be between 4 and 80 years of age and have a GNE-related disorder confirmed by genetic testing.
During their first visit, participants will have the following tests:
- Muscle strength and endurance tests, including walking
- Medical history, physical exam and a neurological exam
- Questionnaires about the impact of the disease on daily activities and quality of life
- Blood and urine sampling
- Heart and lung function tests
- Imaging study of the leg muscles
Participants may return for follow-up visits every 6-12 months. Not all tests will be performed at each visit. Treatment will not be provided as part of this protocol.
For more information, view our Frequently Asked Questions about the study.
What is ManNAc?
ManNAc is an abbreviation for N-acetyl-D-mannosamine. ManNAc is an uncharged sugar and an intermediate molecule in the production of sialic acid within cells (see figure below). Patients with GNE Myopathy have decreased GNE enzyme activity, and do not produce enough ManNAc or sialic acid. This deficiency results in a decreased attachment of sialic acid groups to skeletal muscle cells, which is thought to be the cause of the muscle decline in GNE Myopathy.
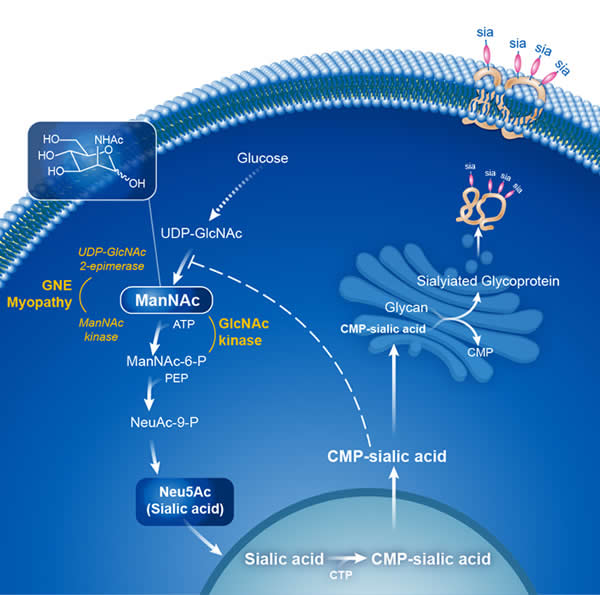
Studies have shown that supplementation of ManNAc to cell cultures or to mice with a dysfunctional GNE protein (as in GNE Myopathy) bypasses the need for GNE function and enables the production of sialic acid. Studies in mouse models of GNE Myopathy have shown a treatment effect of oral ManNAc in mice with GNE Myopathy.
NIH researchers are participating in a multi-center pivotal trial to evaluate the efficacy of ManNAc in GNE Myopathy.
The FDA has provided an orphan designation for ManNAc to treat GNE Myopathy.
Frequently Asked Questions
Where will I have to go to participate in the study?
You will be admitted to the NIH Clinical Center for this study.
Will I get paid to participate in this study?
You will not receive any direct compensation for your participation.
Will I have to pay for my travel expenses to the clinical study center?
Travel expenses will be paid by the NIH. For international patients, we will pay travel from point-of-entry to the US.
Can I bring a companion? Would his/her travel will be paid for?
Yes, if medically indicated, we will pay for a companion.
What are the benefits of participating in this study?
By participating in this study, you will help to improve our understanding of GNE Myopathy and related diseases, which is an important contribution to families and individuals affected by these conditions. This study will provide information that will help all clinical trials that explore new therapies.
Does participating in the GNE Myopathy and GNE-Related Diseases Natural History Study mean that I am automatically enrolled in the ManNAc trials?
No it does not. The eligibility criteria for the ManNAc trial are different from those for this Natural History Study.
Should I stop taking any medications while I participate in this study?
No, we will record all medications taken and analyze the data separately, if needed.
I have multiple family members with GNE Myopathy, can we all participate?
Yes! As long as each person has evidence of a diagnosis of GNE Myopathy and signs informed consent, you may all participate. Furthermore, we can coordinate to bring you and your siblings or friends to the study site at the same time.
How do we participate?
You simply need to email or call us if you are interested in participating: GNEMyopathyStudies@mail.nih.gov
Or contact:
Andrea I. Bowling, CRNP
Research Nurse Practitioner
andrea.bowling@nih.gov
(301) 827-7746
Caitlin Yuan, RN
Research Nurse
caitlin.yuan@nih.gov
(240) 506-4920
Jessica M. Jang, M.P.H.
Clinical Research Coordinator
jessica.jang@nih.gov
(240) 506-4706
We will need:
- Medical records, including a neurology note and a muscle biopsy report (if available)
- Genetic testing results confirming the diagnosis of GNE Myopathy
How do I obtain genetic testing?
Genetic testing is ordered by your physician when he or she suspects that you have GNE Myopathy. Here is information on how to obtain genetic testing: https://www.ncbi.nlm.nih.gov/gtr/conditions/C1853926/
Where will the study take place?
At the National Institutes of Health Clinical Center in Bethesda, MD. For more information, visit the NIH Clinical Center's Patient Information site.
Where can I get more information about GNE Myopathy?
Related Clinical Studies
Ongoing and Recruiting:
- Natural History Study of GNE Myopathy and related disorders
Upcoming Studies:
- Multicenter Clinical Trial of ManNAc for GNE Myopathy
Coming in 2022
Completed Studies
Additional Resources
News Releases
NIH Launches Trial to Evaluate Drug for Rare Degenerative Muscle Disease Treatment (2012)
Mouse Model Points to Possible New Strategy for Treating Rare Muscle Disease, Kidney Disorders (2007)
Lectures by NIH Investigators
GNE Myopathy: Two Decades of Progress – Much Accomplished, Much Still to Do
Presented September 2020: GNEM Symposium Speakers Series
Presenter: Nuria Carrillo, M.D., NIH.
NIH Research Studies for GNE Myopathy
Presented May 2020: GNEM Symposium Speakers Series
Presenter: Marjan Huizing, Ph.D., NHGRI, NIH.
Disorders of Sialic Acid Synthesis: Pathway and Prospects for Therapy
Presented February 2016: SBP Rare Disease Day Symposium 2016
Presenter: Marjan Huizing, Ph.D., NHGRI, NIH.
Advancing ManNAs as a Therapy for GNE Myopathy
Presented February 2015: SBP Rare Disease Day Symposium 2015
Presenter: Nuria Carrillo, M.D., NIH.
N-acetylmannosamine (ManNAc) or Sialic Acid as Therapy for Disorders of Hyposialylation
Presented February 2012: SBP Rare Disease Day Symposium 2012
Presenter: Marjan Huizing, Ph.D., NHGRI, NIH.
Scientific Publications
2021
Carrillo N, Malicdan MC, Leoyklang P, Shrader JA, Joe G, Slota C, Perreault J, Heiss JD, Class B, Liu C-Y, Bradley K, Jodarski C, Ciccone C, Driscoll C, Parks R, Van Wart S, Bayman L, Coffey CS, Quintana M, Berry SM, Huizing M, Gahl WA. Oral N-acetylmannosamine (ManNAc) rescues the basic defect in subjects with GNE myopathy. Genetics in Medicine, accepted (June 2021).
Van Wart SA, Mager DE, Bednasz CJ, Huizing M, Carrillo N. Population Pharmacokinetic Model of N-Acetyl-mannosamine (ManNAc) and N-acetylneuraminic acid (Neu5Ac) in Subjects with GNE Myopathy. Drugs in R&D 2021; Apr 24. doi: 10.1007/s40268-021-00343-6. Online ahead of print. [PubMed]
2020
Carrillo N, Malicdan MC, Huizing M. GNE Myopathy. 2004 Mar 26 [Updated 2020 Apr 9]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. [GeneReviews]
Fang M, Xu X, Zhang M, Shi Y, Gu G, Liu W, Class B, Ciccone C, Gahl WA, Huizing M, Carrillo N, Wang AQ. Quantitation of N-acetylmannosamine, N-acetylneuraminic acid and cytidine-5’-monophospho-N-acetylneuraminic acid in human leukocytes using LC-MS/MS: method development and validation. Biochemical Chromatography 2020; 34: e4735. [PubMed]
2019
Pogoryelova O, Urtizberea JA, Argov Z, Nishino I, Lochmüller H; ENMC workshop study group. "237th ENMC International Workshop: GNE myopathy - current and future research. Hoofddorp, The Netherlands, 14-16 September 2018. Neuromuscular Disorders, 2019; 29: 401-410. [PubMed]
Quintana M, Shrader J, Slota C, Joe G, McKew JC, Fitzgerald M, Gahl WA, Berry S, Carrillo N. Bayesian model of disease progression in GNE myopathy. Statistics in Medicine, 2019 Apr 15;38(8):1459-1474. [PubMed]
2018
Carrillo N, Malicdan MC, Huizing M. GNE Myopathy: Etiology, Diagnosis, and Therapeutic Challenges. Neurotherapeutics. 2018 Oct 18. ;15(4):900-914. [PubMed]
Leoyklang P, Class B, Noguchi S, Gahl WA, Carrillo N, Nishino I, Huizing M, Malicdan MC. Quantification of lectin fluorescence in GNE myopathy muscle biopsies. Muscle Nerve, 2018 Aug;58(2):286-292. [PubMed]
Slota C, Bevans M, Yang L, Shrader J, Joe G, Carrillo N. Patient reported outcomes in GNE myopathy: incorporating a valid assessment of physical function in a rare disease. Disabil Rehabil, 2018 May;40(10):1206-1213. [PubMed]
2017
Garland, J., J. Stephen, B. Class, A. Gruber, C. Ciccone, A. Poliak, C. P. Hayes, V. Singhal, C. Slota, J. Perreault, R. Gavrilova, J. A. Shrader, P. Chittiboina, G. Joe, J. Heiss, W. A. Gahl, M. Huizing, N. Carrillo and M. C. V. Malicdan. Identification of an Alu element-mediated deletion in the promoter region of GNE in siblings with GNE myopathy. Mol Genet Genomic Med, 5(4):410-417. 2017. [PubMed]
Xu, X., A. Q. Wang, L. L. Latham, F. Celeste, C. Ciccone, M. C. Malicdan, B. Goldspiel, P. Terse, J. Cradock, N. Yang, S. Yorke, J. C. McKew, W. A. Gahl, M. Huizing and N. Carrillo. Safety, pharmacokinetics and sialic acid production after oral administration of N-acetylmannosamine (ManNAc) to subjects with GNE myopathy. Mol Genet Metab, April 26, 2017. [PubMed]
2015
Shi Y, Xu X, Fang M, Zhang M, Li Y, Gillespie B, Yorke S, Yang N, McKew JC, Gahl WA, Huizing M, Carrillo-Carrasco N, Wang AQ. Quantitative hydrophilic interaction chromatography-mass spectrometry analysis of N-acetylneuraminic acid and N-acetylmannosamine in human plasma. Journal of Chromatography B, Analytical Technologies in the Biomedical Life Sciences, 000: 105-111. 2015. [PubMed]
Nishino I, Carrillo-Carrasco N, Argov Z. GNE myopathy: current update and future therapy. J Neurol Neurosurg Psychiatry, 86:385-392. 2015. [PubMed]
Hinderlich S, Weidemann W, Yardeni T, Horstkorte R, Huizing M. UDP-GlcNAc 2-epimerase/ ManNAc kinase (GNE): A master regulator of sialic acid synthesis. Topics in Current Chemistry, 366:97-137. 2015. [PubMed]
2014
Huizing M, Malicdan MC, Krasnewich DM, Manoli I, Carrillo-Carrasco N. GNE Myopathy. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G. eds. OMMBID - The Online Metabolic and Molecular Bases of Inherited Diseases. New York: McGraw-Hill; 2014.
de Dios JK, Shrader JA, Joe GO, McClean JC, Williams K, Evers R, Malicdan MC, Ciccone C, Mankodi A, Huizing M, McKew JC, Bluemke DA, Gahl WA, Carrillo-Carrasco N. Atypical presentation of GNE myopathy with asymmetric hand weakness. Neuromuscular Disorders, 24: 1063-1067. 2014. [PubMed]
Celeste F, Vilboux T, Ciccone C, de Dios JK, Malicdan MC, Leoyklang P, McKew JC, Gahl WA, Carrillo-Carrasco N, Huizing M. Mutation Update for GNE Gene Variants Associated with GNE Myopathy. Human Mutation, 35: 915-926. 2014. [PubMed]
Leoyklang P, Malicdan MC, Yardeni T, Celeste F, Ciccone C, Li X, Jiang R, Gahl WA, Carrillo-Carrasco N, He M, Huizing M. Sialylation of Thomsen-Friedenreich antigen is a noninvasive blood-based biomarker for GNE myopathy. Biomarkers in Medicine, 8:641-652. 2014. [PubMed]
Huizing M, Carrillo-Carrasco N, Malicdan MC, Noguchi S, Gahl WA, Mitrani-Rosenbaum S, Argov Z, Nishino I. GNE myopathy: new name and new mutation nomenclature. Neuromuscular Disorders, 24: 387-389. 2014. [PubMed]
Patzel KA, Yardeni T, Le Poëc-Celic E, Leoyklang P, Dorward H, Alonzi DS, Kukushkin NV, Xu B, Zhang Y, Sollogoub M, Blériot Y, Gahl WA, Huizing M, Butters TD. Non-specific accumulation of glycosphingolipids in GNE myopathy. Journal of Inherited Metabolic Disease, 37:297-308. 2014. [PubMed]
2013
Yardeni T, Jacobs K, Niethamer TK, Ciccone C, Anikster Y, Kurochkina N, Gahl WA, Huizing M. Murine isoforms of UDP-GlcNAc 2-epimerase/ManNAc kinase: Secondary structures, expression profiles, and response to ManNAc therapy. Glycoconjugate Journal, 30:609-618. 2013. [PubMed]
2012
Niethamer TK, Yardeni T, Leoyklang P, Ciccone C, Astiz-Martinez A, Jacobs K, Dorward HM, Zerfas PM, Gahl WA, Huizing M. Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Molecular Genetics and Metabolism, 107:748-755. 2012. [PubMed]
Kakani S, Yardeni T, Poling J, Ciccone C, Niethamer T, Klootwijk RD, Manoli I, Darvish D, Hoogstraten-Miller S, Zerfas P, Tian E, Ten Hagen KG, Kopp JB, Gahl WA, Huizing M. The Gne M712T mouse as a model for human glomerulopathy. The American Journal of Pathology , 180:1431-1440. 2012. [PubMed]
2011
Nemunaitis G, Jay CM, Maples PB, Gahl WA, Huizing M, Yardeni T, Tong AW, Phadke AP, Pappen BO, Bedell C, Allen H, Hernandez C, Templeton NS, Kuhn J, Senzer N, Nemunaitis J. Hereditary Inclusion Body Myopathy: Single Patient Response to Intravenous Dosing of GNE Gene Lipoplex. Human Gene Therapy, 22:1331-1341. 2011. [PubMed]
Yardeni T, Choekyi T, Jacobs K, Ciccone C, Patzel K, Anikster Y, Gahl WA, Kurochkina N, Huizing M. Identification, Tissue Distribution and Molecular Modeling of Novel Human Isoforms of the Key Enzyme in Sialic Acid Synthesis, UDP-GlcNAc 2-epimerase/ManNAc Kinase. Biochemistry, 50:8914-8925. 2011. [PubMed]
2010
Nemunaitis G, Maples PB, Jay C, Gahl WA, Huizing M, Poling J, Tong AW, Phadke AP, Pappen BO, Bedell C, Templeton NS, Kuhn J, Senzer N, Nemunaitis J. Hereditary Inclusion Body Myopathy: Single Patient Response to GNE Gene Lipoplex Therapy. The Journal of Gene Medicine, 12:403-412. 2010. [PubMed]
Voermans NC, Guillard M, Doedée R, Lammens M, Huizing M, Padberg GW, Wevers RA, van Engelen BG, Lefeber DJ. Clinical features, lectin staining, and a novel GNE frameshift mutation Hereditary Inclusion Body Myopathy. Clinical Neuropathology, 29:71-77. 2010. [PubMed]
Kurochkina N, Yardeni T, Huizing M. Molecular modeling of the bifunctional enzyme uridine diphosphate-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase and predictions of structural effects of mutations associated with HIBM and sialuria. Gycobiology, 20:322-337. 2010. [PubMed]
2009
Huizing M, Krasnewich DM. Hereditary Inclusion Body myopathy: A decade of progress. Biochimica et Biophysica Acta, Molecular Basis of Disease, 1792:881-887. 2009. [PubMed]
2007
Galeano B, Klootwijk R, Manoli I, Sun MS, Ciccone C, Darvish D, Starost MF, Zerfas PM, Hoffmann VJ, Hoogstraten-Miller S, Krasnewich DM, Gahl WA, Huizing M. N-Acetylmannosamine treatment rescues UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase knock-in mice from severe neonatal glomerular hematuria and podocytopathy. Journal of Clinical Investigation, 17:1585-1594. 2007. [PubMed]
Sparks S, Rakocevic G, Joe G, Manoli I, Shrader J, Sonies B, Ciccone C, Dorward H, Krasnewich D, Huizing M, Dalakas M, Gahl W. Pilot Study of the Use of Intravenous Immune Globulin in Hereditary Inclusion Body Myopathy. BMC Neurology, 7:3. [PubMed]
2006
Savelkoul PJM, Manoli I, Sparks S, Ciccone C, Gahl WA, Krasnewich DM, Huizing M. Normal sialylation status of N-linked and O-GalNAc linked glycans in Hereditary Inclusion Body Myopathy. Molecular Genetics and Metabolism, 88:389-390. 2006. [PubMed]
2005
Sparks SE, Ciccone C, Lalor M, Orvisky E, Klootwijk R, Savelkoul PJ, Dalakas MC, Krasnewich DM, Gahl WA, Huizing M. Use of a cell-free system to determine UDP-N-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase activities in human Hereditary Inclusion Body Myopathy. Glycobiology, 15:1102-1110. 2005. [PubMed]
Gottlieb E, Ciccone C, Darvish D, Naiem S, Dalakas MC, Savelkoul PJ, Krasnewich DM, Gahl WA, Huizing M. Single nucleotide polymorphisms within the dystroglycan gene in hereditary inclusion body myopathy. Molecular Genetics and Metabolism, 86: 244-249. 2005. [PubMed]
2004
Huizing M, Rakocevic G, Sparks SE, Mamali I, Shatunov A, Goldfarb L, Krasnewich D, Gahl WA, Dalakas M. Hypoglycosylation of α-dystroglycan in patients with hereditary IBM due to GNE mutations. Molecular Genetics and Metabolism, 81:196-202. 2004. [PubMed]
GNE Myopathy Team
Francis Rossignol, M.D.
Principal Investigator of GNE Myopathy Clinical Studies at NIH
francis.rossignol@nih.gov
Dr. Rossignol follows one of the largest cohorts of national and international patients with GNE Myopathy at the NIH Clinical Center. He is interested in understanding the natural history of the disease and advancing promising therapies for GNE Myopathy.
Marjan Huizing. Ph.D.
Staff Scientist
mhuizing@mail.nih.gov
Dr. Huizing studies disease mechanisms of GNE Myopathy since 2001. Her team's studies on a mouse model of GNE Myopathy identified that oral ManNAc therapy could increase tissue sialylation in a mouse model of GNE Myopathy. She has been involved in all stages of clinical development of ManNAc for GNE Myopathy at NIH.
May Christine Malicdan, M.D., Ph.D.
Staff Scientist
maychristine.malicdan@nih.gov
Dr. Malicdan joined the NIH research team in 2011. She studies disease mechanism and biomarker development of GNE Myopathy. In 2009, Dr. Malicdan was the lead author of a landmark paper describing sialylation-increasing therapies in a mouse model of GNE Myopathy, which completed as a graduate student in Dr. Nishino's laboratory (Tokyo, Japan).
William A. Gahl, M.D., Ph.D.
Director, NIH Undiagnosed Diseases Program
gahlw@mail.nih.gov
Dr. Gahl's laboratory has a longstanding interest in sialic acid metabolism and its associated human disorders. Dr. Gahl initiated clinical studies of GNE Myopathy at NIH, including a pilot clinical trial with highly sialylated IVIg (in 2005). He also directed and sponsored the first trials of ManNAc in GNE Myopathy.
Petcharat Leoyklang, Ph.D.
Biologist
petcharat.leoyklang@nih.gov
Carla Ciccone, M.Sc.
Biologist, Laboratory Manager
cciccone@mail.nih.gov
Contact Information
New Patients
To reach the GNE Myopathy Study team: GNEMyopathyStudies@mail.nih.gov.
Or contact the Clinical Team:
Andrea I. Bowling, CRNP
Research Nurse Practitioner
andrea.bowling@nih.gov
(301) 827-7746
Caitlin Yuan, RN
Research Nurse
caitlin.yuan@nih.gov
(240) 506-4920
Jessica M. Jang, MPH
Clinical Research Coordinator
jessica.jang@nih.gov
(240) 506-4706
For Basic Research
Marjan Huizing, Ph.D.
mhuizing@mail.nih.gov
(240) 893-4742
To Submit Medical Records
Fax: 301-402-0006
eFax: 301-451-6769
Address
NIH Clinical Center
10 Center Drive
Building 10, Room 10C103
Bethesda, MD 20895
USA
Last updated: January 28, 2022