
Human Genomic Variation
The vast majority of the DNA letters in peoples’ genomes is identical, but a small fraction of those letters varies. This genomic variation accounts for some of the differences among people, including important aspects of their health and susceptibility to diseases.
The Big Picture
-
Genomic variation reflects the differences in a person’s DNA compared to other peoples’ DNA.
-
There are multiple types of variants in human genomes, ranging from small differences to large differences.
-
A very small subset of genomic variants contributes to human health and disease.
-
Researchers create reference human genome sequences to help detect genomic variants in each sequenced human genome.
-
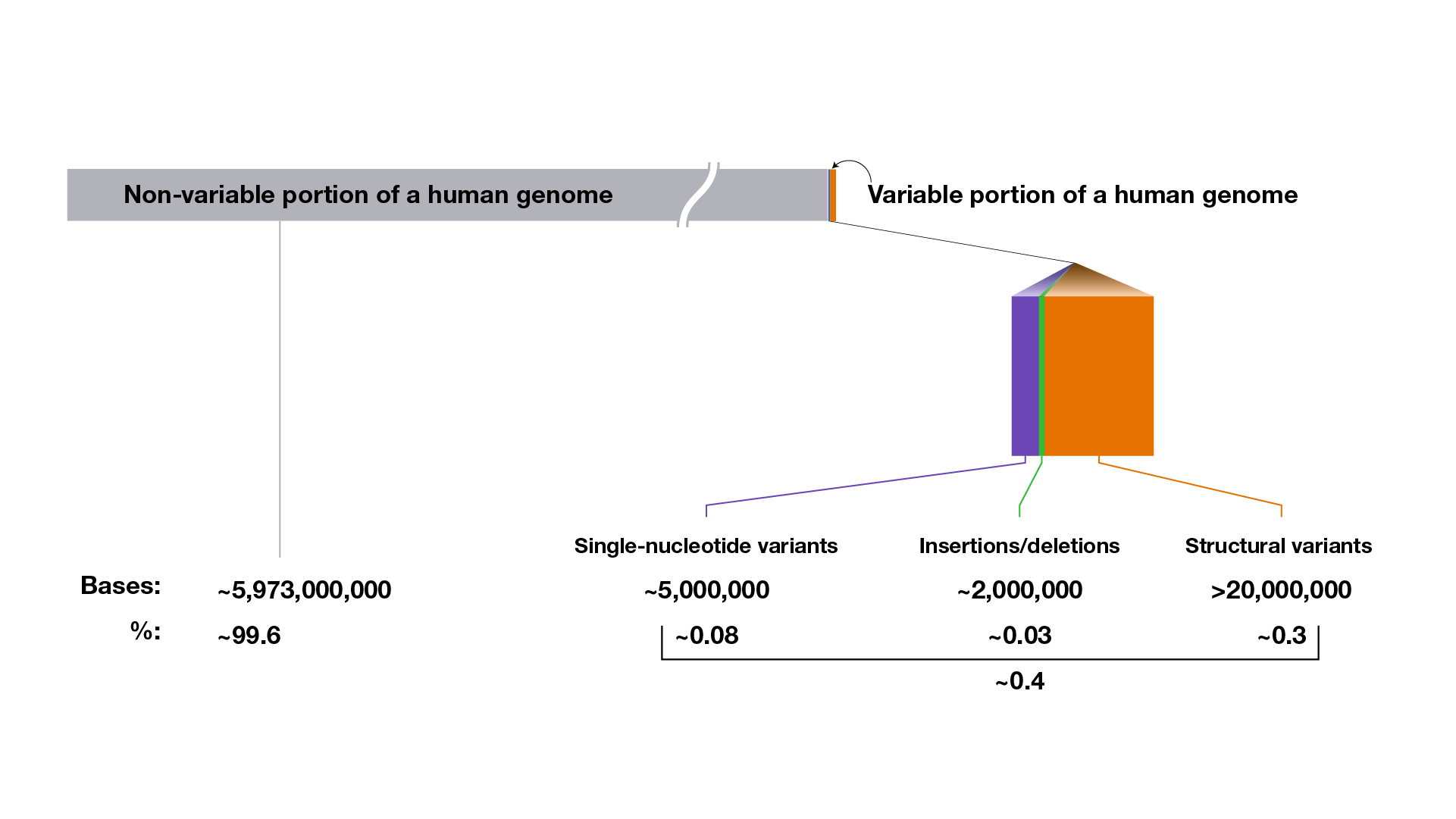
On average, a person’s genome sequence is ~99.6% identical to a reference human genome sequence; that person’s set of genomic variants accounts for the ~0.4% difference.
-
The human pangenome is a more comprehensive framework that aims to account for genomic variation across human populations, thereby reducing biases that can come with the use of a single reference human genome sequence.
What is the human genome?
A genome is the complete set of DNA instructions found in every cell. DNA is made of four different chemicals (called nucleotides or bases), each represented by a different letter: adenine (A), thymine (T), cytosine (C) and guanine (G). The order of these letters (i.e., the DNA sequence) encodes the information that instructs each cell what to do and when to do it. The genome’s DNA is packed into structures called chromosomes.
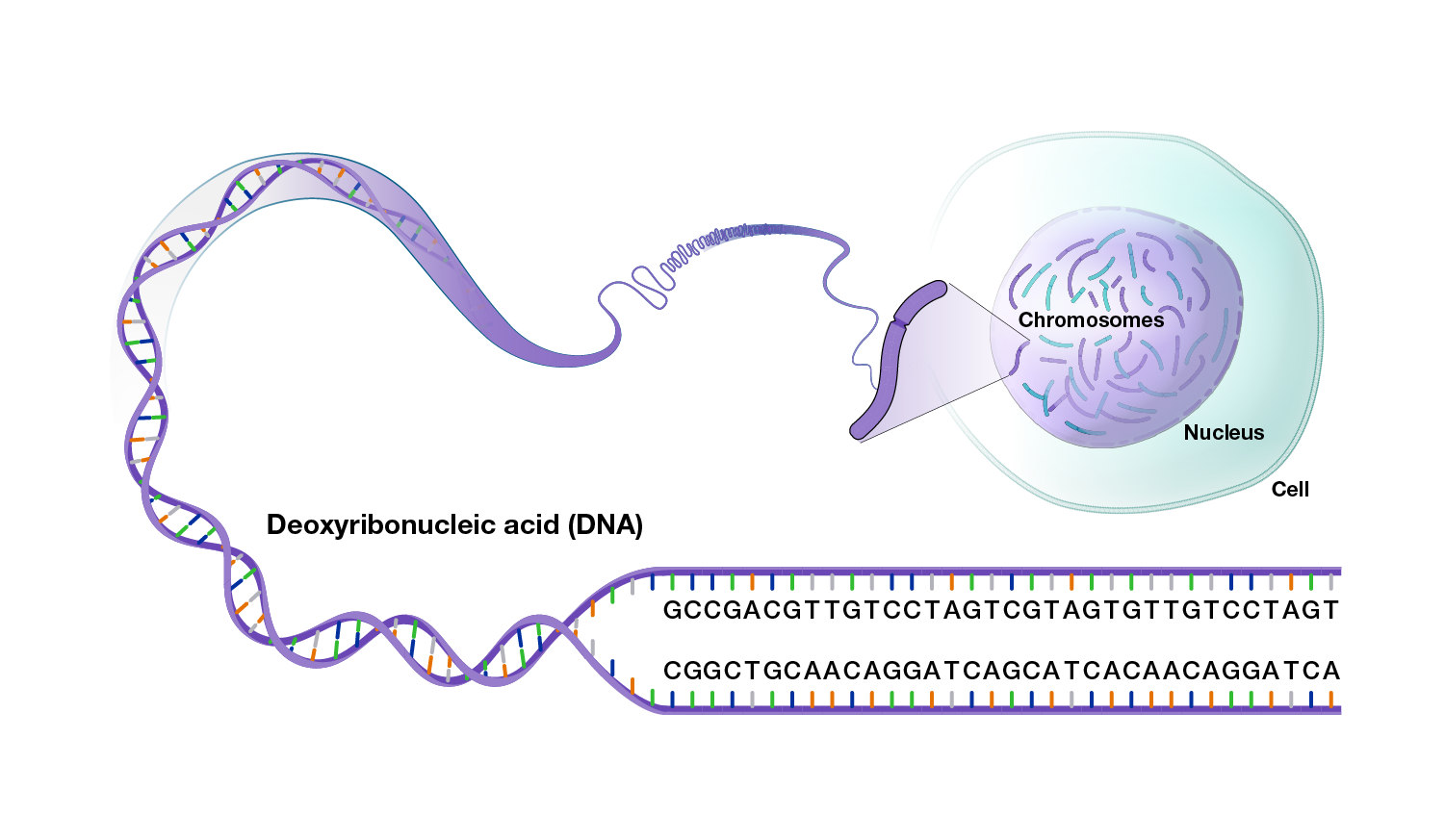
One copy of the human genome contains about 3 billion nucleotides, which are distributed among 23 chromosomes. Most human cells have two copies of the human genome, with one copy inherited from each parent. Cells containing two copies of each chromosome are called “diploid.” Most mammals are diploid, but some organisms have either one set or more than two sets of each chromosome.
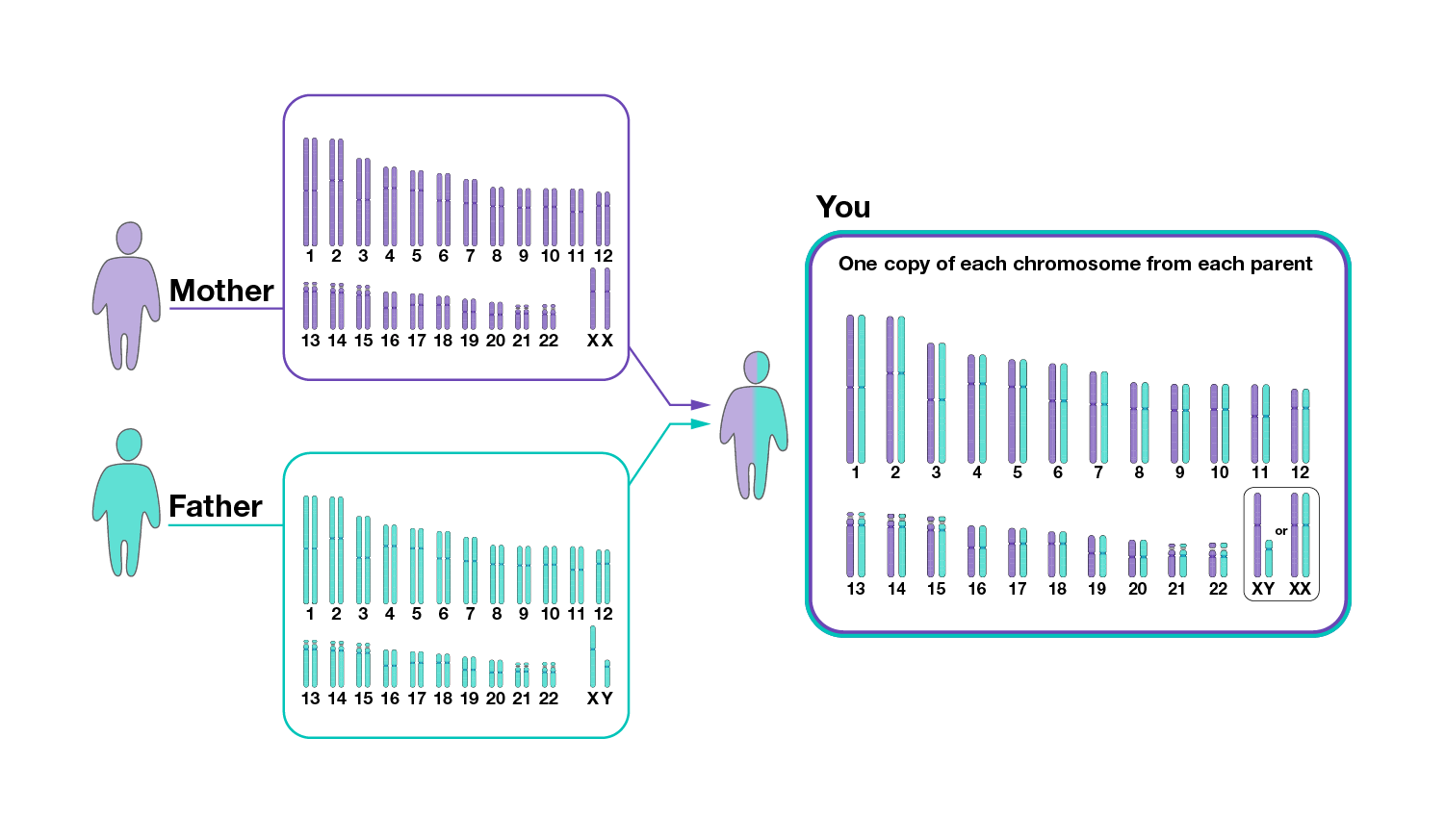
How do peoples’ genomes vary?
Peoples’ genomes are far more similar to each other than they are different. It is frequently stated that any two peoples’ genomes are ~99.9% identical to one another. This percentage is based on the finding that, on average, a single-nucleotide difference exists between two peoples’ genomes once every 1,300 nucleotides or so.
However, this is an over-simplification because it only accounts for single-nucleotide differences. In reality, any two peoples’ genomes are, on average, ~99.6% identical and ~0.4% different. The latter percentage reflects both single-nucleotide differences and differences that involve multiple nucleotides (discussed in detail below).
The differences among human genomes are called genomic variants. A person’s set of genomic variants is part of what makes them unique. Other factors (such as diet, environment, lifestyle and social context) also contribute to a person’s uniqueness. Most genomic variants have no influence on the functioning of a person’s genome, but a small subset of variants do have an impact. For example, some genomic variants influence physical characteristics, like eye color and height; others influence health conditions or how a person responds to certain medications.
What are the different types of genomic variants?
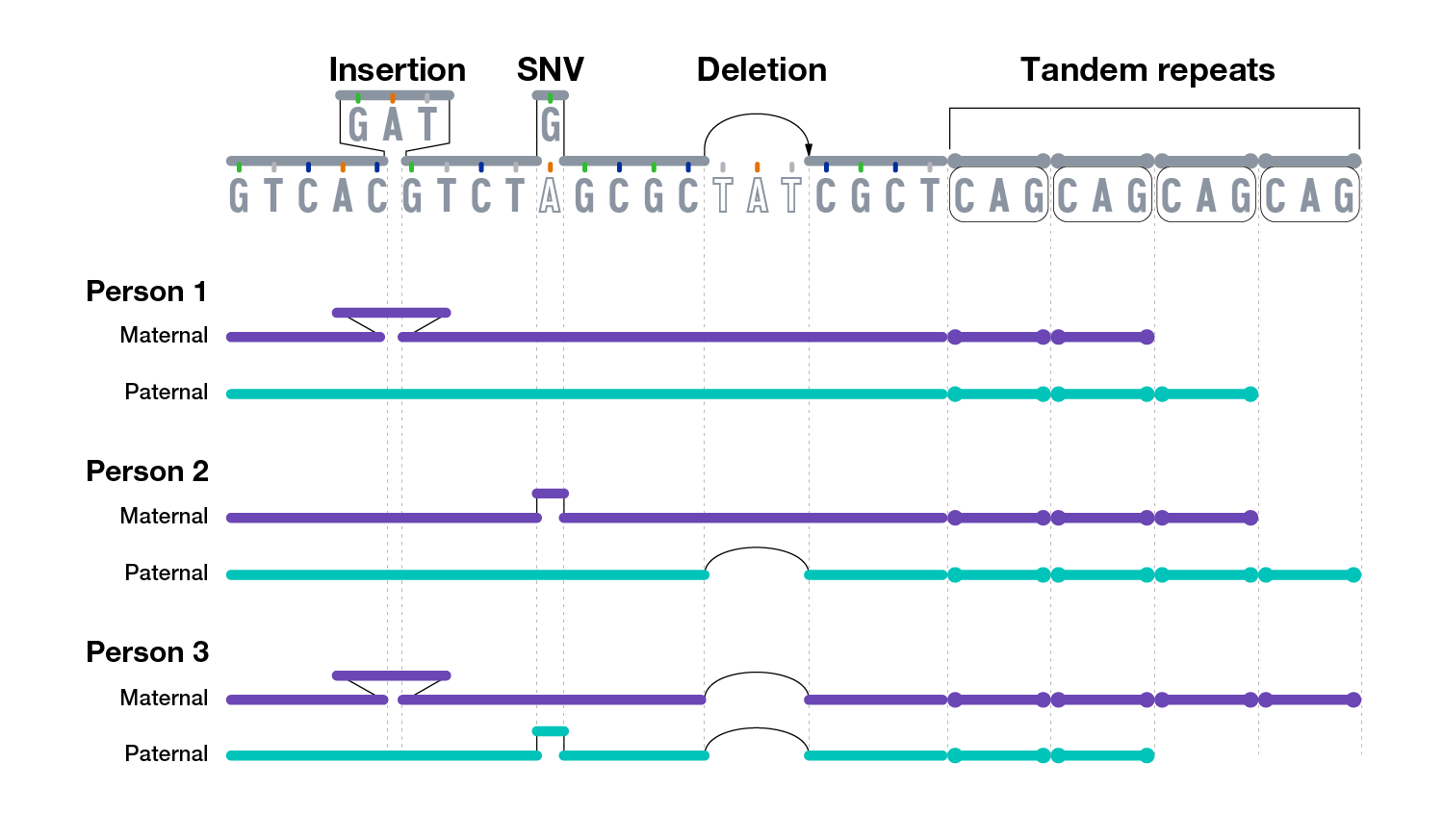
There are multiple types of genomic variants.
The smallest genomic variants are single-nucleotide variants (SNVs). Each SNV reflects a difference in a single nucleotide (or letter). For a given SNV, the DNA letter at that genomic position might be a C in one person but a T in another person. SNVs are the most common type of genomic variation. A subtype of SNVs is called a single-nucleotide polymorphism (SNP; pronounced “snip”). To be considered a SNP, a SNV must be present in at least 1% of the human population. As such, SNV is a more general term that includes both relatively common (such as SNPs) and rare single-nucleotide differences. For simplicity, we refer to all single-nucleotide differences as SNVs, regardless of their relative frequency.

Another group of small genomic variants are insertions and deletions (often referred to as “indels”). Insertion/deletion variants reflect extra or missing DNA nucleotides in the genome, respectively, and typically involve fewer than 50 nucleotides. Insertion/deletion variants are less frequent than SNVs but can sometimes have a larger impact on health and disease (e.g., by disrupting the function of a gene that encodes an important protein).
One of the most common types of insertion/deletion variants are tandem repeats (also known as microsatellites). Tandem repeats are short stretches of nucleotides that are repeated multiple times and are highly variable among people. Different chromosomes can vary in the number of times such short nucleotide stretches are repeated, ranging from a few times to hundreds of times. Historically, tandem repeats have been used for building maps of the human genome and DNA profiling in forensics applications.
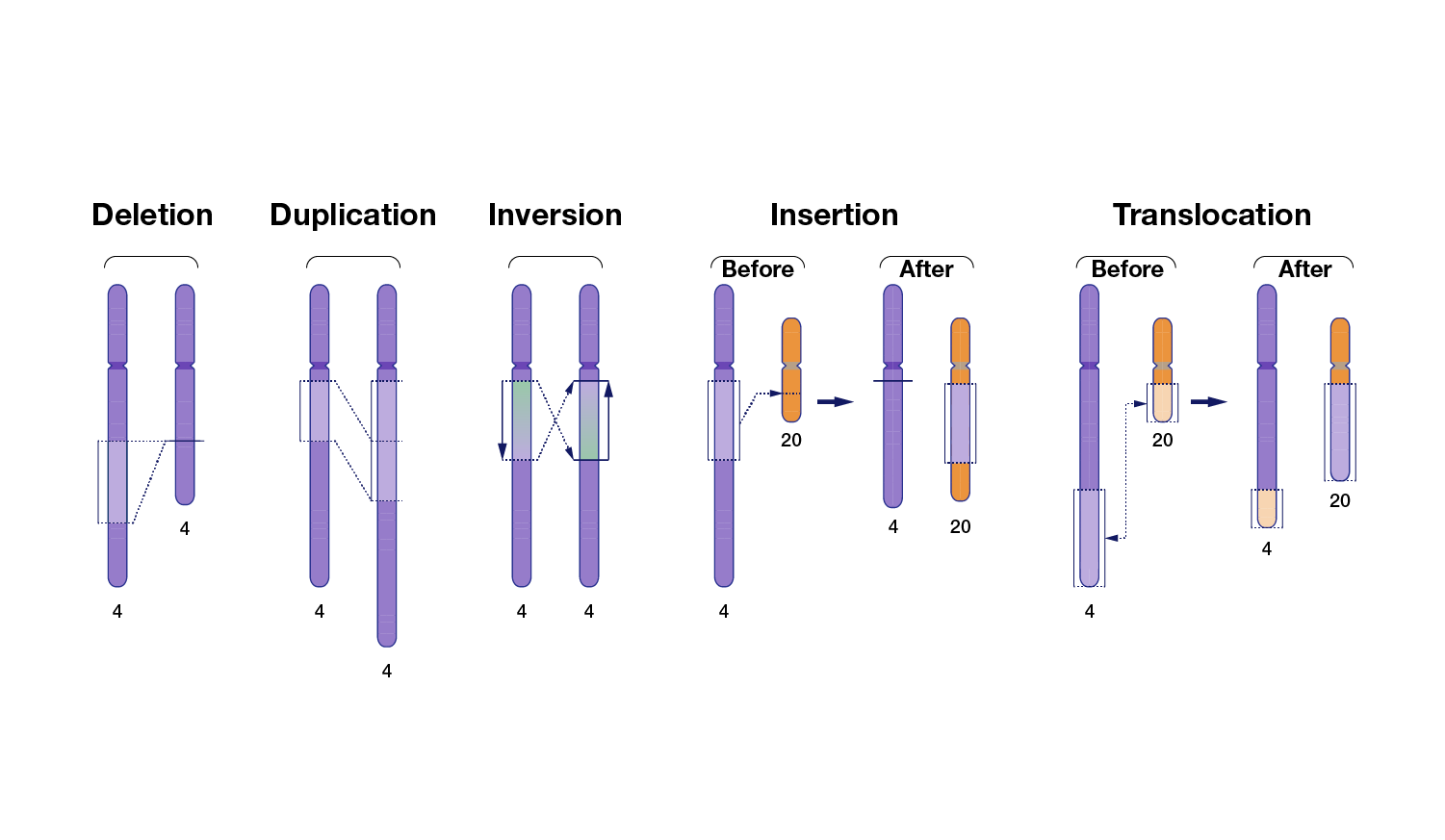
Genomic variation also extends beyond small stretches of nucleotides to larger chromosomal regions. These large-scale genomic differences are called structural variants and involve at least 50 nucleotides and as many as thousands of nucleotides that have been inserted, deleted, inverted or moved from one part of the genome to another. Tandem repeats that contain more than 50 nucleotides are considered structural variants; in fact, such large tandem repeats account for nearly half of the structural variants present in human genomes. When a structural variant reflects differences in the total number of nucleotides involved, it is called a copy-number variant (CNV). Note that CNVs are distinguished from other structural variants, such as inversions and translocations, because the latter types often do not involve a difference in the total number of nucleotides.
How are genomic variants detected?
Researchers use different approaches to detect genomic variants.
Some methods are designed to only detect known genomic variants. For example, it might be important to know if a person has inherited a particular genomic variant that is relevant to their health or healthcare; for that, researchers can perform a specific DNA test to determine whether a person has that genomic variant. With other methods, researchers can analyze a person’s DNA for the presence of a large number of known genomic variants; for example, a type of test called a microarray can detect hundreds of thousands of SNVs at once.
A more comprehensive way to detect genomic variants, including those that might not yet be known, is to perform genome sequencing. Multiple methods are now available for sequencing human genomes, each only requiring a small sample of blood, hair or cheek cells from which DNA is isolated. Today, sequencing a human genome usually costs less than $1,000, which is over a million-fold less expensive than a couple of decades ago.
When a person’s genome is sequenced, both copies of each pair of chromosomes (one from each parent) are sequenced at the same time. In most routine situations (e.g., when a genome is sequenced for a medical purpose), it is not readily possible to determine the parent of origin for each detected genomic variant. This is because a human genome is not sequenced one chromosome at a time; rather, the process involves breaking up the entire genome and then piecing back together small stretches of the sequenced DNA, like a jigsaw puzzle. Without additional analyses, that process does not provide information about the parental origin of each genomic variant. A “reference” human genome sequence is also critical to sequencing a human genome and detecting genomic variants.
What is a reference genome sequence?
Detecting variants in a person’s genome sequence usually requires an existing sequence for comparison — a “reference.” A reference human genome sequence is an established, high-quality and well-accepted sequence of a human genome (i.e., one sequence for each of the 23 human chromosomes). A reference human genome sequence is not an actual sequence from one individual but pieced together from multiple people. The important feature of a reference human genome sequence is that it depicts one assigned nucleotide for every position across the human genome. In this regard, a reference human genome sequence only represents the sequence of one copy of each chromosome, whereas a person’s genome sequence contains the sequences of both copies of each chromosome (i.e., people are diploid).

Of course, the human population contains an immense amount of variation among genomes. A given reference human genome sequence is not intended to capture or represent that diversity; rather, it serves as an important data-analysis tool. Specifically, each newly generated genome sequence from a person is directly compared to a given reference human genome sequence, allowing for the detection of all differences (variants) between the person’s genome and the reference genome. In short, comparison of a person’s genome sequence to a reference genome sequence allows for the cataloguing of all genomic variants in that person’s genome.
What is the inventory of genomic variants in a typical human genome sequence?
What does a typical human genome sequence look like with respect to the variants that it contains? On average, compared to a reference human genome, a person’s ~6 billion-nucleotide genome sequence will have:
-
~5,000,000 SNVs that involve ~5,000,000 nucleotides
-
~600,000 insertion/deletion variants that involve ~2,000,000 nucleotides
-
~25,000 structural variants that involve >20,000,000 nucleotides.
These numbers represent current estimated averages and will likely change as more human genomes are sequenced to completion. Furthermore, multiple approaches can be used to calculate how the different types of genomic variants account for the differences among genomes. In fact, calculating the total number of nucleotides involved in structural variants in each human genome is quite complicated and remains an active area of research.
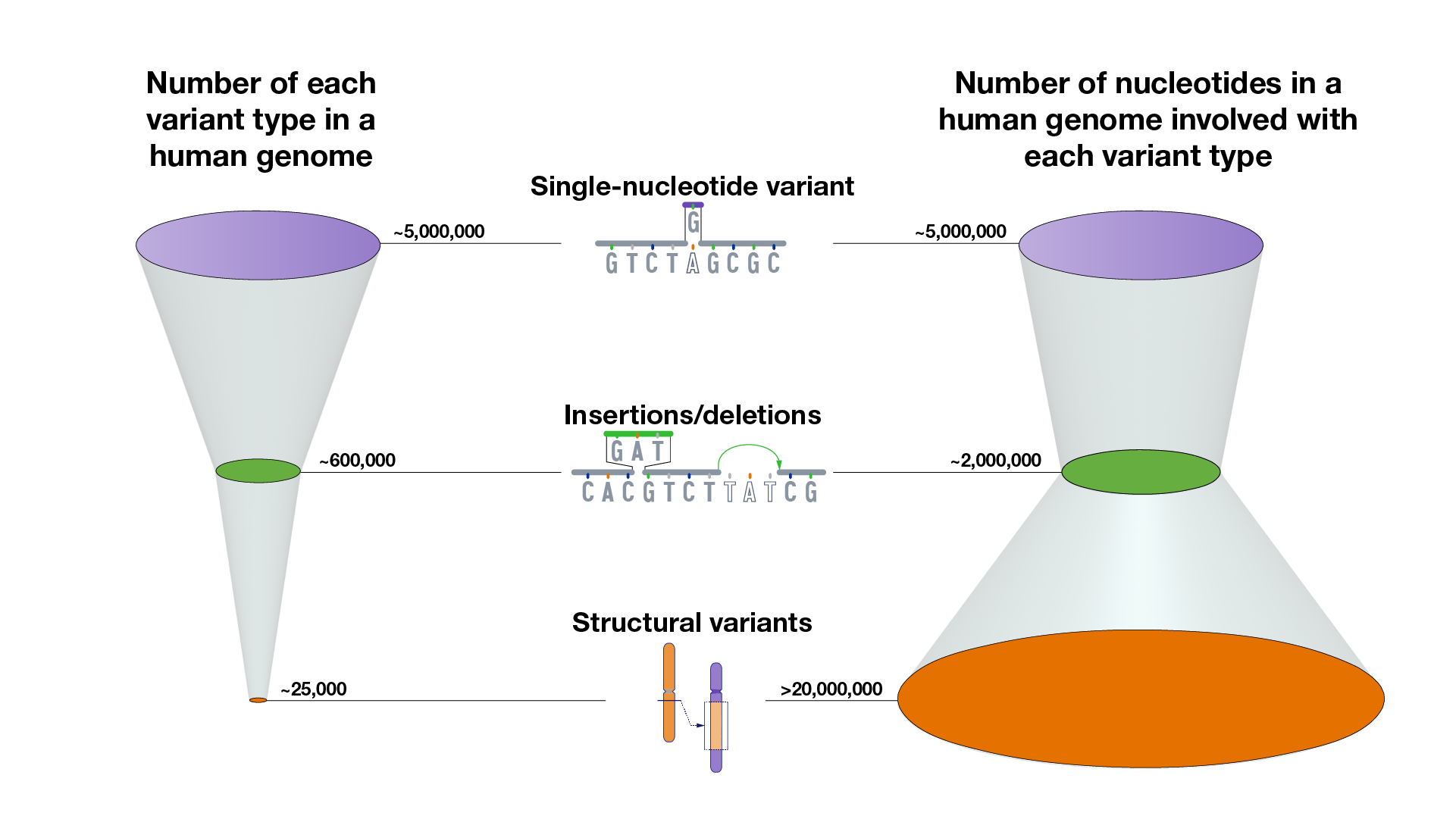
That means that, on average, the complete set of genomic variants in each person’s genome involves ~27,000,000 nucleotides (among the ~6,000,000,000 nucleotides in their genome). Those ~27,000,000 nucleotides reflect some type of difference at those positions in the DNA, together accounting for ~0.4% of the person’s complete genome. In other words, when accounting for the full inventory of genomic variants, a typical person’s genome sequence is ~99.6 identical to (or ~0.4% different from) a reference human genome sequence (or even another person’s genome sequence).
What is a pangenome reference sequence and how is it used?
Reference human genome sequences are invaluable for detecting genomic variants in each newly sequenced human genome. But the currently available reference human genome sequences do not accurately represent the genomic diversity of the human population, and that lack of diversity can introduce biases when analyzing some peoples’ genome sequences.
To address this deficiency, researchers are working to generate a more complete set of reference human genome sequences that better reflect all of humanity. A “pangenome” is the collective genome sequences of multiple individuals that better represents the genomic diversity of the species. The human pangenome reference sequence will provide a better tool for comparing genome sequences from people all over the world in an effort to detect and characterize genomic variants more completely, including those with important roles in health and disease.
Looking forward
Genomics is constantly evolving and advancing, including the regular development of new experimental technologies and data-analysis methods. Such advances are readily applicable to ongoing efforts to develop new and more inclusive sets of reference human genome sequences and improve the ability to detect all variants in each newly sequenced human genome. The long-term goal is to have sufficient knowledge about genomic variation in all human populations, bringing equity in the benefits of genomic medicine.


Last updated: February 1, 2023