

Cynthia Tifft, M.D., Ph.D.
Office Of The Clinical Director
Medical Genetics Branch
Glycosphingolipid and Glycoprotein Disorders Unit
B.A. University of California, San Diego
M.S. Rutgers University
Ph.D. University of Texas, Houston
M.D. University of Texas, Houston
Biography
Dr. Tifft is a senior clinician at the National Human Genome Research Institute where she serves as deputy clinical director and directs the Pediatric Undiagnosed Diseases Program.
Dr. Tifft received her B.A. in biology from Revelle College at the University of California at San Diego and her M.S. in genetic counseling from Rutgers University. She received her Ph.D. in genetics from the University of Texas Graduate School of Biomedical Science at M.D. Anderson Cancer Center and her M.D. from the University of Texas Health Science Center in Houston. She completed her pediatric residency at Johns Hopkins Hospital and fellowship in medical genetics at the National Institutes of Health. Dr. Tifft joined the faculty of the George Washington University School of Medicine at Children's National Medical Center in 1991, becoming chair of the Division of Genetics and Metabolism in 1996. In 2009, she returned to the NIH to focus her attention on children with rare and undiagnosed diseases.
For many years, Dr. Tifft's clinical and research interests have been lysosomal disorders affecting the central nervous system. She is the principal investigator of a longitudinal natural history study of children and adults with glycosphingolipid and glycoprotein disorders, including Tay-Sachs and Sandhoff diseases, GM1 gangliosidosis and type 1 sialidosis.
In May 2019 Dr. Tifft and her “all-woman team from five continents” launched the initial first-in-human gene therapy trial for patients with Type I and Type II GM1 gangliosidosis. Connecting with desperate families and enrolling patients in the middle of a pandemic gave participants the chance at a better life. For their stellar work the team received the NHGRI GREAT Award in 2020 and the NIH Director’s Award in 2021.

As director of the Pediatric Undiagnosed Diseases Program, Dr. Tifft coordinates the selection and clinical evaluation of children whose diagnoses have long eluded the medical community. Using comprehensive phenotyping and next generation sequencing, she and her team have provided diagnoses to nearly a third of selected patients and have strong candidate genes under study for additional cases.
GM1 Gangliosidosis
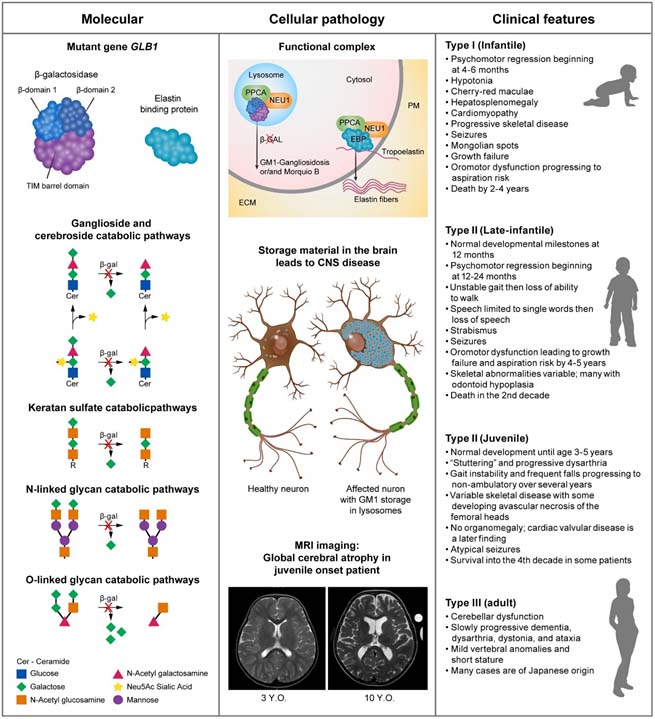
Toro C, Zainab M, Tifft CJ. The GM2 gangliosidoses: Unlocking the mysteries of pathogenesis and treatment. Neurosci Lett. 2021 Nov 1;764:136195. doi: 10.1016/j.neulet.2021.136195. Epub 2021 Aug 25. PMID: 34450229; PMCID: PMC8572160.
GM2 Gangliosidosis
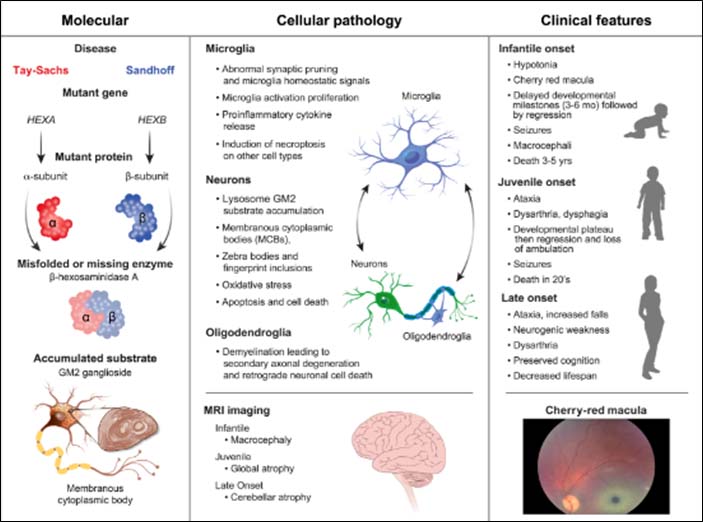
Nicoli ER, Annunziata I, d'Azzo A, Platt FM, Tifft CJ, Stepien KM. GM1 Gangliosidosis-A Mini-Review. Front Genet. 2021 Sep 3;12:734878. doi: 10.3389/fgene.2021.734878. PMID: 34539759; PMCID: PMC8446533.
Glycosphingolipid Disorders and Glycoprotein Disorders Unit

- Research Nurse Coordinator
- Office of the Clinical Director

- Pediatric Nurse Practitioner
- NIH Undiagnosed Diseases Program

- Postbaccalaureate Fellow
- Glycosphingolipid and Glycoprotein Disorders Unit

- Postbaccalaureate Fellow
- Glycosphingolipid Disorders and Glycoprotein Disorders Unit



Last updated: January 12, 2025